Scalable Preparation of High-Purity 3'-O-Methoxyethyl Nucleosides for Antisense Drug Development
Introduction to Advanced Nucleoside Modification Technology
The rapid evolution of genomic medicine has placed antisense oligonucleotide (ASO) drugs at the forefront of therapeutic innovation, driven by their superior specificity and reduced side effect profiles compared to traditional small molecules. Central to the development of second-generation ASOs is the availability of high-quality modified nucleoside building blocks, specifically 3'-O-methoxyethyl nucleosides, which enhance binding affinity and nuclease resistance. Patent CN108424432B, published in April 2021, discloses a groundbreaking preparation method that addresses the critical industrial bottlenecks associated with synthesizing these compounds. Unlike previous literature reports that struggled with low yields and difficult separation of isomers, this novel approach utilizes a robust synthetic route starting from readily available diacetone allose. By implementing a strategic sequence of alkylation, oxidation-reduction, and selective protection, the process achieves exceptional stereocontrol, ensuring the exclusive formation of the desired 3'-substituted product while effectively suppressing the generation of 2'-isomers. This technological leap provides a reliable 3'-O-methoxyethyl nucleoside supplier pathway that meets the stringent purity requirements essential for clinical-grade oligonucleotide manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 3'-O-methoxyethyl nucleosides has been plagued by significant chemical challenges that hindered their transition from laboratory curiosities to commercial commodities. Traditional routes often resulted in complex mixtures containing both 2'- and 3'-substituted isomers, which possess nearly identical physical properties, making their separation an arduous task. In many prior art methods, the removal of these isomers necessitated the use of preparative high-performance liquid chromatography (HPLC) or repeated recrystallization cycles, both of which are economically prohibitive at an industrial scale. Preparative chromatography suffers from low throughput and high solvent consumption, while repeated crystallization inevitably leads to substantial yield losses, rendering the overall process inefficient. Furthermore, existing methods frequently relied on starting materials that were either expensive or difficult to source in bulk, creating supply chain vulnerabilities. The inability to consistently achieve high purity without specialized equipment meant that the cost reduction in antisense oligonucleotide manufacturing remained elusive, limiting the widespread adoption of these promising therapeutic agents.
The Novel Approach
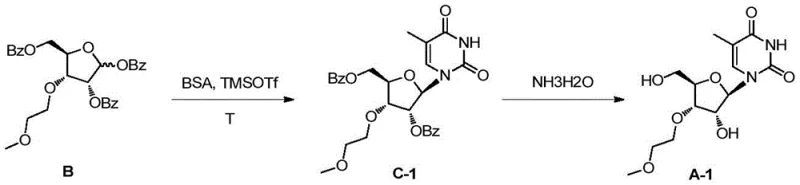
The methodology outlined in the patent represents a paradigm shift by introducing a highly convergent and stereoselective synthetic strategy that circumvents the isomerization issues inherent in older techniques. The core innovation lies in the construction of a specific key intermediate, Formula B, derived from 1,2:5,6-diacetone-D-allose through a series of controlled transformations. This intermediate features a unique protection pattern where the 1, 2, and 5-hydroxyl positions are benzoylated, creating a steric environment that directs the subsequent glycosylation reaction exclusively to the 3'-position. The process employs mild and scalable reagents, such as trimethylsilyl trifluoromethanesulfonate (TMSOTf) or stannic chloride, to catalyze the condensation with silylated bases like thymine, 5-methylcytosine, or adenine. Crucially, the final deprotection and purification steps are streamlined; the target nucleosides can be isolated via simple crystallization from ethanol, achieving purities exceeding 99.5% without the need for chromatographic separation. This approach not only simplifies the operational workflow but also drastically improves the overall yield and economic viability of producing high-purity nucleoside intermediates.

Mechanistic Insights into Stereoselective Glycosylation and Sugar Modification
The chemical elegance of this synthesis is rooted in the precise manipulation of carbohydrate stereochemistry and protecting group orthogonality. The transformation begins with the regioselective alkylation of the primary hydroxyl group on the diacetone allose scaffold using 2-chloroethyl methyl ether under basic conditions (50% KOH in DMSO), which installs the methoxyethyl moiety with high fidelity. Following this, the 5,6-acetonide protecting group is selectively removed using trifluoroacetic acid (TFA), exposing the vicinal diol necessary for the subsequent carbon-chain shortening. The oxidative cleavage of this diol using sodium periodate (NaIO4) generates an aldehyde intermediate, which is immediately reduced by sodium borohydride (NaBH4) to yield the ribose-configured alcohol (Formula IM4). This oxidation-reduction sequence is critical as it effectively converts the allose backbone into the desired ribose structure while retaining the 3'-methoxyethyl modification. The mechanistic control continues into the protection phase, where benzoyl chloride is used to cap the remaining hydroxyl groups. The resulting tribenzoylated sugar (Formula B) acts as a rigid glycosyl donor that, upon activation by a Lewis acid, forms an oxocarbenium ion intermediate. The bulky benzoyl groups shield the 2'-position, forcing the silylated nucleobase to attack from the beta-face at the 1'-position, thereby locking in the correct anomeric configuration and preventing the formation of alpha-anomers or 2'-linked byproducts.
Impurity control is inherently built into this molecular design, addressing the most critical concern for R&D directors focused on drug safety. The use of benzoyl protecting groups serves a dual purpose: it enhances the lipophilicity of the intermediate, facilitating easier extraction and purification, and it provides the necessary steric bulk to enforce regioselectivity during the coupling reaction. In conventional syntheses, the lack of such directing groups often leads to a statistical distribution of isomers that are notoriously difficult to separate. Here, the specific electronic and steric properties of the tribenzoylated intermediate ensure that the condensation reaction proceeds with high specificity. Furthermore, the final alkaline hydrolysis step using concentrated ammonia water is gentle enough to remove the benzoyl groups without degrading the sensitive glycosidic bond or the methoxyethyl ether linkage. This robustness allows for the production of 3'-O-methoxyethyl nucleosides with impurity profiles that are significantly cleaner than those obtained via traditional routes, reducing the burden on downstream processing and ensuring that the commercial scale-up of complex modified sugars remains feasible and compliant with regulatory standards.
How to Synthesize 3'-O-Methoxyethyl Nucleosides Efficiently
The synthesis of these valuable nucleoside analogs follows a logical progression from abundant carbohydrate feedstocks to highly functionalized pharmaceutical intermediates. The process initiates with the alkylation of 1,2:5,6-diacetone-D-allose to introduce the methoxyethyl side chain, followed by a sequence of deprotection, oxidation, and reduction to establish the ribose core. Subsequent benzoylation creates the activated glycosyl donor, which is then coupled with a protected nucleobase under Lewis acid catalysis. The detailed standardized synthetic steps, including specific reagent ratios, temperature controls, and workup procedures required to replicate this high-yielding process, are outlined below.
- Alkylate 1,2: 5,6-diacetone-D-allose with 2-chloroethyl methyl ether under basic conditions to introduce the methoxyethyl group.
- Perform selective deprotection, periodate oxidation, and borohydride reduction to convert the allose derivative into the ribose configuration.
- Protect hydroxyl groups with benzoyl chloride, then condense with a silylated base using TMSOTf or SnCl4 catalyst followed by alkaline hydrolysis.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the implementation of this patented technology offers transformative benefits that directly impact the bottom line and operational resilience. The primary advantage lies in the drastic simplification of the purification process; by eliminating the need for preparative chromatography, manufacturers can significantly reduce solvent consumption, waste disposal costs, and processing time. This shift from low-throughput separation techniques to high-efficiency crystallization enables a substantial increase in production capacity without requiring capital-intensive equipment upgrades. Moreover, the reliance on 1,2:5,6-diacetone-D-allose as a starting material leverages a commodity chemical that is widely available and cost-effective, insulating the supply chain from the volatility often associated with specialized fine chemical precursors. The robustness of the reaction conditions, which do not require cryogenic temperatures or exotic catalysts, further enhances the reliability of supply, ensuring consistent delivery schedules for downstream oligonucleotide synthesis.
- Cost Reduction in Manufacturing: The elimination of preparative HPLC and the reduction in crystallization cycles lead to significant operational savings. By avoiding yield losses associated with repeated purification steps, the overall material efficiency is greatly improved, translating to a lower cost per gram of the final active ingredient. Additionally, the use of common reagents like benzoyl chloride and ammonia water minimizes raw material expenses compared to processes requiring specialized protecting group reagents.
- Enhanced Supply Chain Reliability: The use of stable, commercially available starting materials reduces the risk of supply disruptions. The synthetic route is designed to be scalable, allowing for seamless transition from pilot plant to multi-ton production scales. This scalability ensures that procurement teams can secure long-term contracts with confidence, knowing that the supplier has the technical capability to meet increasing demand as antisense therapies move through clinical trials to market.
- Scalability and Environmental Compliance: The process operates under relatively mild conditions and avoids the use of hazardous heavy metal catalysts in the majority of steps (except for specific adenine couplings where SnCl4 is an option, but TMSOTf is preferred for others), aligning with modern green chemistry principles. The simplified workup procedures reduce the volume of organic waste generated, facilitating easier compliance with environmental regulations and lowering the costs associated with waste treatment and disposal.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of 3'-O-methoxyethyl nucleosides. These answers are derived directly from the experimental data and process descriptions found in the patent literature, providing a transparent view of the technology's capabilities and limitations for potential partners and stakeholders.
Q: How does this method avoid the formation of 2'-O-methoxyethyl isomers?
A: The method utilizes a specific protection strategy on the D-ribose intermediate (Formula B) where the 1, 2, and 5 positions are benzoylated. This steric environment, combined with the use of specific Lewis acid catalysts like TMSOTf during the condensation step, directs the nucleophilic attack of the base specifically to the 3'-position, effectively suppressing the formation of the unwanted 2'-isomer.
Q: What is the primary starting material for this synthesis?
A: The synthesis begins with 1,2:5,6-diacetone-D-allose (Formula SM), which is a cheap and commercially abundant carbohydrate. This contrasts with other methods that might require more expensive or less available sugar precursors, significantly aiding in cost reduction for large-scale manufacturing.
Q: Is preparative chromatography required for purification?
A: No, one of the key advantages of this patented process is that the final product (Formula A) can be purified via simple crystallization from ethanol. This eliminates the need for expensive and low-throughput preparative chromatography, making the process highly suitable for industrial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3'-O-Methoxyethyl Nucleoside Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful development of next-generation antisense drugs depends on the availability of ultra-high purity building blocks produced via scalable and robust methodologies. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive a consistent supply of materials that meet stringent purity specifications. Our state-of-the-art rigorous QC labs are equipped to analyze complex nucleoside impurity profiles, guaranteeing that every batch of 3'-O-methoxyethyl nucleoside delivered adheres to the highest quality standards required for GMP manufacturing. We are committed to bridging the gap between innovative academic research and industrial reality, providing the technical expertise necessary to navigate the complexities of modified sugar synthesis.
We invite pharmaceutical companies and research institutions to collaborate with us to accelerate their drug development programs. By leveraging our optimized synthesis platforms, we can offer a Customized Cost-Saving Analysis tailored to your specific project needs, identifying opportunities to further optimize the supply chain. We encourage you to contact our technical procurement team today to request specific COA data for our nucleoside portfolio and to discuss route feasibility assessments for your custom synthesis requirements, ensuring a partnership built on quality, reliability, and scientific excellence.