Advanced Chiral Synthesis of Anticoagulant Intermediates via Amidoxime Reduction
Advanced Chiral Synthesis of Anticoagulant Intermediates via Amidoxime Reduction
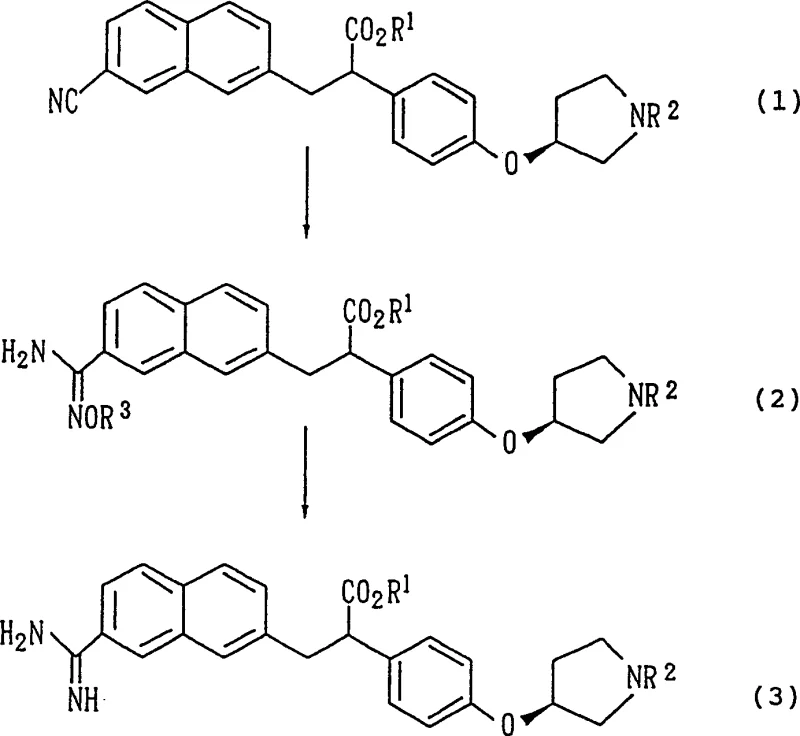
The pharmaceutical industry's relentless pursuit of safer and more effective anticoagulants has placed significant emphasis on the efficient synthesis of thrombin inhibitor intermediates. Patent CN1181053C introduces a groundbreaking methodology for preparing 3-(7-amidino-2-naphthyl)-2-phenylpropionic acid derivatives, which serve as critical precursors for potent antithrombotic agents. Unlike conventional routes that struggle with stereochemical integrity, this invention leverages a robust amidoxime intermediate strategy to ensure exceptional optical purity. For R&D directors and process chemists, this represents a pivotal shift from hazardous, low-yielding ammonolysis reactions to a controlled, two-step sequence involving hydroxylamine addition followed by selective reduction. The technical implications extend beyond mere yield improvements; they address the fundamental challenge of maintaining chiral fidelity in complex aromatic systems, thereby offering a reliable pathway for the commercial scale-up of complex pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of aromatic amidine derivatives relied heavily on direct ammonolysis of nitrile precursors, a process fraught with significant operational and chemical drawbacks. As detailed in the background of the patent, traditional methods necessitate the use of corrosive hydrogen chloride gas and anhydrous ammonia, creating severe safety hazards and requiring specialized containment infrastructure that drives up capital expenditure. Furthermore, to suppress unwanted epimerization at the chiral alpha-carbon, these reactions must be conducted at extremely low temperatures for extended periods, often exceeding one week. This prolonged exposure to acidic conditions inevitably leads to partial racemization, as evidenced by prior art where optical purity dropped from 99.7% de to roughly 94.8% de. Such degradation is unacceptable for modern regulatory standards, where tight control over impurity profiles and enantiomeric excess is mandatory for API approval. Additionally, the presence of acid-labile protecting groups, such as tert-butyloxycarbonyl, often results in premature deprotection and byproduct formation, complicating downstream purification and reducing overall process efficiency.
The Novel Approach
In stark contrast, the methodology disclosed in CN1181053C circumvents these pitfalls by introducing a discrete amidoxime intermediate, fundamentally altering the reaction landscape to favor stereochemical retention. This novel approach replaces the harsh ammonolysis conditions with a mild nucleophilic addition of hydroxylamine to the nitrile group, followed by a gentle reduction step. By decoupling the nitrogen introduction from the final amidine formation, the process allows for precise control over reaction parameters, significantly shortening the total cycle time from weeks to merely a few hours. The use of common solvents like ethanol and readily available reagents such as hydroxylamine sulfate simplifies the supply chain logistics and reduces raw material costs. Most critically, this route demonstrates remarkable stability of the chiral center, with experimental data showing that optical purity is maintained at approximately 99.1% de throughout the transformation. This preservation of stereochemistry eliminates the need for costly chiral resolution steps later in the synthesis, providing a direct cost reduction in pharmaceutical intermediate manufacturing while ensuring a consistent, high-quality supply of the active scaffold.
Mechanistic Insights into Amidoxime Formation and Catalytic Reduction
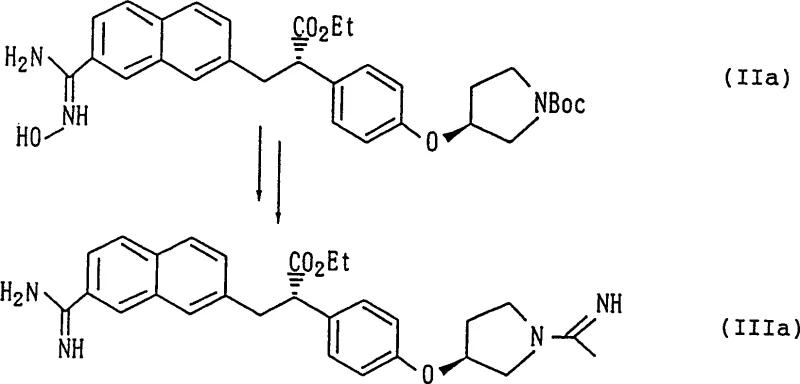
The core of this technological advancement lies in the mechanistic elegance of the two-step transformation, which begins with the nucleophilic attack of hydroxylamine on the electrophilic carbon of the nitrile group. In the first stage, the nitrile compound reacts with hydroxylamine or its salts, such as hydroxylamine hydrochloride or sulfate, typically in an alcoholic solvent like ethanol. The reaction proceeds through the formation of an imidate intermediate which tautomerizes to the more stable amidoxime structure. This step is kinetically favorable and can be driven to completion by refluxing the mixture for 1 to 6 hours, a drastic improvement over the week-long timelines of previous methods. The choice of solvent is crucial; protic solvents facilitate the proton transfer necessary for the rearrangement, while the moderate basicity generated in situ (often by neutralizing the hydroxylamine salt with sodium hydroxide) ensures rapid conversion without degrading sensitive functional groups elsewhere in the molecule. The resulting amidoxime is a robust intermediate that can often be isolated and purified via crystallization, acting as a checkpoint to remove unreacted starting materials before the final reduction.
The second mechanistic phase involves the selective reduction of the amidoxime N-O bond to yield the target amidine, a transformation that requires careful catalyst selection to avoid over-reduction or side reactions. The patent highlights the efficacy of transition metal catalysts, specifically palladium on carbon (Pd/C), which facilitates hydrogenation under mild conditions. Alternatively, transfer hydrogenation using formic acid serves as a safe and effective hydrogen source, eliminating the need for high-pressure hydrogen gas equipment. The reduction mechanism likely involves the adsorption of the amidoxime onto the metal surface, followed by the cleavage of the weak oxygen-nitrogen bond and subsequent protonation to form the C-N double bond of the amidine. Crucially, these conditions are sufficiently mild to prevent the base-catalyzed or acid-catalyzed racemization that plagues direct ammonolysis. By avoiding strong mineral acids during the reduction phase and utilizing buffered conditions or weak organic acids, the configuration at the chiral benzylic position remains intact. This mechanistic control is the key to achieving the high optical purity observed in the examples, ensuring that the final API precursor meets the stringent enantiomeric specifications required for anticoagulant therapeutics.
How to Synthesize 3-(7-amidino-2-naphthyl) Derivatives Efficiently
The practical implementation of this synthesis route offers a streamlined protocol for producing high-value anticoagulant intermediates with minimal environmental impact and maximum safety. The process begins by dissolving the chiral nitrile precursor in ethanol and treating it with an aqueous solution of hydroxylamine sulfate neutralized with sodium hydroxide. The mixture is heated to reflux for a short duration, typically around 2 hours, until TLC analysis confirms the complete consumption of the nitrile starting material. Upon cooling, the amidoxime intermediate often precipitates out of the solution, allowing for simple filtration and washing to obtain a pure solid. This solid is then subjected to reduction, either by stirring with Pd/C and formic acid at room temperature or by using zinc dust in an acidic medium. The reaction progress is monitored to ensure full conversion to the amidine, after which the catalyst is removed by filtration. The final product is isolated as a stable salt, such as the dihydrochloride or maleate, through crystallization from appropriate solvent systems.
- React the chiral nitrile compound with hydroxylamine or its salt in an alcoholic solvent (e.g., ethanol) under reflux to form the amidoxime intermediate.
- Perform catalytic hydrogenation or metal-mediated reduction (using Pd/C or Zinc/Iron) on the amidoxime to generate the target amidine compound.
- Isolate the final product as a stable salt (e.g., dihydrochloride or maleate) through crystallization to ensure high optical purity and pharmaceutical grade quality.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this patented process translates into tangible strategic benefits that extend far beyond the laboratory bench. The elimination of hazardous gaseous reagents like anhydrous ammonia and hydrogen chloride significantly lowers the barrier to entry for manufacturing partners, as it removes the requirement for specialized high-pressure reactors and extensive scrubbing systems. This simplification of the equipment footprint directly correlates to reduced capital investment and lower operational overheads, making the supply chain more resilient and less prone to regulatory shutdowns due to safety compliance issues. Furthermore, the drastic reduction in reaction time from over a week to merely a few hours dramatically increases plant throughput, allowing manufacturers to respond more agilely to market demand fluctuations without needing to expand facility capacity. The ability to isolate stable intermediates also provides flexibility in production scheduling, enabling batch staging that optimizes resource utilization across the manufacturing site.
- Cost Reduction in Manufacturing: The economic impact of this process is profound, primarily driven by the removal of expensive and dangerous reagents and the associated safety infrastructure. By replacing high-pressure ammonolysis with ambient pressure hydroxylamine reactions, the process eliminates the need for costly corrosion-resistant alloys and complex gas handling systems, leading to substantial cost savings in equipment maintenance and operation. Additionally, the high yield and minimal byproduct formation reduce the burden on waste treatment facilities and lower the consumption of solvents and purification media. The preservation of optical purity throughout the synthesis negates the need for expensive chiral chromatography or resolution steps, which are often the most cost-prohibitive part of asymmetric synthesis. Consequently, the overall cost of goods sold (COGS) for the intermediate is significantly lowered, providing a competitive pricing advantage in the global API market.
- Enhanced Supply Chain Reliability: Supply continuity is bolstered by the use of commodity chemicals such as ethanol, hydroxylamine salts, and palladium catalysts, which are readily available from multiple global suppliers, reducing the risk of single-source bottlenecks. The robustness of the reaction conditions means that the process is less sensitive to minor variations in raw material quality or environmental factors, ensuring consistent batch-to-batch reproducibility. This reliability is critical for long-term supply agreements with major pharmaceutical companies, where deviation in quality can lead to costly delays in drug registration. Moreover, the shorter cycle time allows for faster inventory turnover, reducing the working capital tied up in work-in-progress stock and enabling a more lean and responsive supply chain model that can adapt quickly to changes in downstream API demand.
- Scalability and Environmental Compliance: From an environmental and scaling perspective, this methodology aligns perfectly with modern green chemistry principles and increasingly stringent regulatory frameworks. The avoidance of volatile organic compounds and toxic gases minimizes the facility's emissions profile, simplifying the permitting process for new production lines. The aqueous workup and crystallization steps generate less hazardous waste compared to traditional extraction-heavy processes, lowering disposal costs and environmental liability. Scalability is inherently supported by the exothermic nature of the reactions being manageable under reflux conditions, allowing for safe scale-up from kilogram to multi-ton production without encountering the heat transfer limitations often seen in cryogenic ammonolysis. This makes the process ideal for commercial scale-up of complex pharmaceutical intermediates, ensuring that supply can meet the demands of blockbuster anticoagulant drugs without compromising on safety or sustainability standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology, derived directly from the patent specifications and experimental data. Understanding these nuances is essential for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios. The answers provided reflect the specific advantages demonstrated in the patent examples, particularly regarding stereochemical stability and process safety.
Q: How does this process prevent epimerization compared to traditional ammonolysis?
A: Traditional methods using ammonia and hydrogen chloride gas require prolonged reaction times (over one week) and harsh conditions that promote racemization at the chiral center. This novel amidoxime route operates under milder conditions with significantly shorter reaction times, maintaining optical purity above 99% de.
Q: What catalysts are preferred for the reduction step in this synthesis?
A: The patent specifies palladium catalysts, particularly palladium-on-carbon (Pd/C), as highly effective. Transfer hydrogenation using formic acid as a hydrogen source is also described as a preferred method to avoid high-pressure hydrogen equipment.
Q: Is this method suitable for large-scale industrial production?
A: Yes, the method is explicitly designed for industrial scalability. It eliminates the need for handling large volumes of hazardous hydrogen chloride and ammonia gases, simplifies purification through crystallization, and achieves high yields suitable for commercial API manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-(7-amidino-2-naphthyl) Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-fidelity synthesis in the development of next-generation anticoagulants. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the intricate stereochemical requirements of compounds like those in CN1181053C are met with precision. We operate stringent purity specifications and utilize rigorous QC labs equipped with advanced chiral HPLC capabilities to guarantee that every batch of intermediate delivered meets the exacting standards required for clinical and commercial API manufacture. Our commitment to quality assurance means that we can reliably supply complex chiral building blocks that maintain the optical integrity necessary for potent thrombin inhibition.
We invite potential partners to engage with our technical procurement team to discuss how this optimized synthesis route can be tailored to your specific project needs. By leveraging our expertise in process optimization, we can provide a Customized Cost-Saving Analysis that quantifies the economic benefits of switching to this amidoxime-based methodology. We encourage you to contact us to request specific COA data and route feasibility assessments, allowing you to make informed decisions that enhance both the speed and efficiency of your drug development pipeline.