Advanced One-Step Synthesis of Amlodipine: Enhancing Purity and Commercial Scalability for Global Pharma Supply Chains
Advanced One-Step Synthesis of Amlodipine: Enhancing Purity and Commercial Scalability for Global Pharma Supply Chains
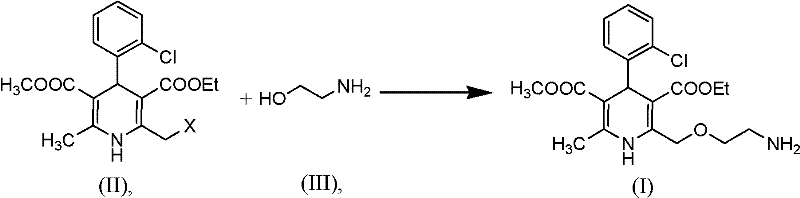
The pharmaceutical landscape for cardiovascular treatments continues to evolve, with amlodipine remaining a cornerstone therapy as a long-acting calcium channel blocker. Patent CN102070516A introduces a transformative methodology for the preparation of this critical active pharmaceutical ingredient (API), addressing long-standing challenges in synthetic efficiency and impurity control. Unlike conventional multi-step sequences that rely on cumbersome protection and deprotection strategies, this invention discloses a direct nucleophilic substitution reaction. By reacting a specific dihydropyridine precursor, designated as Formula (II), with aminoethanol in the presence of a base, manufacturers can achieve high-purity amlodipine in a single operational step. This technical breakthrough not only streamlines the synthetic pathway but also significantly mitigates the risk of introducing persistent organic impurities that compromise drug safety. As a reliable pharmaceutical intermediates supplier, understanding these mechanistic advantages is crucial for optimizing supply chain resilience and product quality.
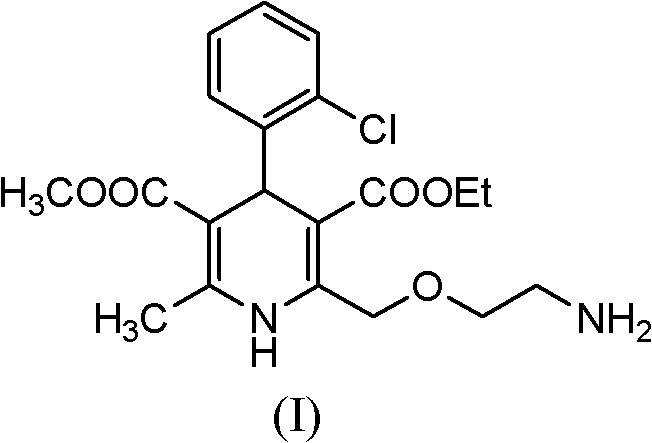
Amlodipine exhibits excellent bioavailability and a prolonged duration of action, making it a first-choice treatment for hypertension and angina. However, the commercial viability of any API is heavily dependent on the robustness of its manufacturing process. The structural complexity of amlodipine, particularly the chiral center and the sensitive dihydropyridine ring, demands precise synthetic control. The method described in CN102070516A leverages a leaving group strategy at the 2-position side chain of the dihydropyridine scaffold. Whether the leaving group X is a halogen such as chlorine or bromine, or a sulfonate ester like mesylate, the reaction proceeds efficiently under mild conditions. This flexibility in substrate selection allows procurement teams to source the most cost-effective precursors without compromising the integrity of the final transformation, thereby supporting cost reduction in pharmaceutical intermediates manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
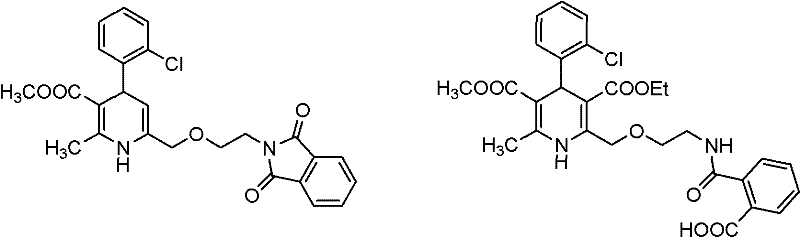
Historically, the industrial synthesis of amlodipine has been plagued by inefficiencies associated with amino protection strategies. Prior art, including various US patents, typically involves the introduction of a protecting group on the amino side chain to prevent unwanted side reactions during the formation of the dihydropyridine ring. While chemically logical, this approach necessitates a subsequent deprotection step, often utilizing hazardous reagents such as hydrazine, azanol, or methylamine. These deprotection conditions are not only dangerous to handle on a large scale but also generate specific degradation products known as Impurity A and Impurity B. These impurities are structurally similar to the target molecule, making them notoriously difficult to remove via standard crystallization or chromatography. Their presence in the final drug substance poses significant regulatory risks, as European pharmacopoeia standards strictly limit impurity content to below 0.3%. Consequently, traditional routes often suffer from lower overall yields and increased production costs due to the extensive purification required to meet safety specifications.
The Novel Approach
The innovative process outlined in CN102070516A circumvents these pitfalls by eliminating the protection-deprotection cycle entirely. Instead of building the molecule with a protected amine and then stripping it away, this method constructs the final side chain directly through a nucleophilic displacement. The precursor, Formula (II), contains a pre-installed leaving group at the terminal position of the side chain. When treated with aminoethanol (Formula III) under basic conditions, the amine nucleophile attacks the electrophilic carbon, displacing the leaving group X and forming the desired ether-amine linkage in one fell swoop. This direct approach drastically reduces the number of unit operations, solvent usage, and reaction time. Furthermore, by avoiding hydrazine-based deprotection, the formation of Impurity A and Impurity B is inherently prevented. This results in a cleaner crude product profile, simplifying downstream processing and ensuring that the final high-purity calcium channel blocker meets stringent regulatory requirements with minimal effort.
Mechanistic Insights into Base-Catalyzed Nucleophilic Substitution
The core of this synthetic advancement lies in the efficient execution of the nucleophilic substitution reaction. The mechanism involves the activation of monoethanolamine by a base to enhance its nucleophilicity, followed by an SN2-type attack on the primary carbon bearing the leaving group. The choice of base is critical; the patent exemplifies the use of strong bases such as sodium hydride, potassium tert-butoxide, or even sodium metal. These bases serve to deprotonate the hydroxyl or amino group of the ethanolamine, or simply to scavenge the acid generated during the displacement (HX), driving the equilibrium toward product formation. The reaction is typically conducted in aprotic polar solvents like tetrahydrofuran (THF), dimethylformamide (DMF), or dimethyl sulfoxide (DMSO), which stabilize the transition state and solubilize the ionic intermediates. Alternatively, weakly nucleophilic alcohols like isopropanol or t-butanol can be used, offering a greener solvent profile. The versatility of the leaving group X allows for optimization based on reactivity and cost; while halides like chlorine are inexpensive, sulfonates like mesylates offer superior leaving group ability, potentially allowing for milder reaction temperatures.
From an impurity control perspective, the mechanism offers inherent selectivity. The dihydropyridine ring is sensitive to oxidation and acidic conditions, but the basic environment of this reaction preserves the integrity of the heterocycle. Moreover, the absence of hydrazine eliminates the risk of hydrazine adducts forming on the ester functionalities of the dihydropyridine ring, a common side reaction in older methods. The patent data indicates that reaction temperatures can be carefully controlled, ranging from -30°C to reflux, with many embodiments successfully operating at room temperature (25°C). This thermal flexibility is a significant advantage for process safety and energy consumption. By fine-tuning the stoichiometry of the base and aminoethanol—typically using a slight excess of 1 to 5 equivalents—manufacturers can ensure complete conversion of the precious dihydropyridine intermediate while minimizing the formation of bis-alkylated byproducts. This precise control over reaction kinetics is essential for achieving the reported purity levels of up to 99.9% for the besylate salt.
How to Synthesize Amlodipine Efficiently
The implementation of this synthesis route requires careful attention to anhydrous conditions and reagent quality to maximize yield and purity. The process begins with the preparation of the nucleophilic species by mixing the base and aminoethanol in a dry solvent, followed by the controlled addition of the dihydropyridine precursor. Detailed standard operating procedures regarding addition rates, temperature ramps, and workup protocols are critical for reproducibility. For a comprehensive guide on the specific experimental parameters, stoichiometry, and isolation techniques validated in the patent examples, please refer to the standardized synthesis steps provided below.
- Prepare the reaction system by suspending a strong base such as sodium hydride or potassium tert-butoxide in an anhydrous aprotic solvent like tetrahydrofuran under inert atmosphere.
- Add monoethanolamine to the base mixture to generate the nucleophilic species, ensuring hydrogen evolution is complete before proceeding to the next stage.
- Introduce the dihydropyridine precursor containing a leaving group at the side chain, maintain reaction temperature between 0°C and room temperature, and isolate the product via extraction and crystallization.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthetic route offers compelling economic and operational benefits. The primary advantage stems from the drastic simplification of the manufacturing workflow. By collapsing multiple steps into a single transformation, the process significantly reduces the consumption of solvents, reagents, and energy. This reduction in material intensity translates directly into lower variable costs per kilogram of produced API. Furthermore, the elimination of hazardous deprotection reagents like hydrazine reduces the burden on waste treatment facilities and lowers the costs associated with environmental compliance and worker safety protocols. The simplified process flow also means shorter cycle times, allowing manufacturing facilities to increase throughput and respond more rapidly to market demand fluctuations without requiring additional capital investment in new reactor trains.
- Cost Reduction in Manufacturing: The economic impact of removing the protection-deprotection sequence cannot be overstated. Traditional routes incur costs not only for the protecting group reagents but also for the additional solvents and energy required for the extra reaction and isolation steps. By bypassing these stages, the novel method achieves a leaner cost structure. Additionally, the high purity of the crude product reduces the need for expensive preparative chromatography or multiple recrystallizations, further driving down the cost of goods sold (COGS). The ability to use inexpensive leaving groups like chlorine instead of specialized sulfonates provides additional flexibility in raw material sourcing, allowing procurement teams to optimize spend based on market availability.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the robustness of this chemistry. The reagents involved—aminoethanol, common bases like potassium tert-butoxide, and solvents like THF or toluene—are commodity chemicals with stable global supply chains. This reduces the risk of production stoppages due to the shortage of exotic or highly regulated reagents. Moreover, the reaction conditions are relatively mild, often proceeding at room temperature, which reduces the dependency on specialized cryogenic equipment or high-pressure reactors. This operational simplicity ensures consistent batch-to-batch quality and reliable delivery schedules, making it easier to qualify as a reliable pharmaceutical intermediates supplier for major generic and branded drug manufacturers.
- Scalability and Environmental Compliance: Scaling chemical processes often introduces new challenges regarding heat transfer and mixing, but this nucleophilic substitution is inherently scalable. The exotherm is manageable, and the use of standard solvents facilitates easy solvent recovery and recycling, aligning with green chemistry principles. The avoidance of genotoxic impurities like hydrazine derivatives simplifies the regulatory filing process and reduces the long-term liability associated with trace contaminants. This environmental and regulatory alignment is increasingly important for multinational corporations aiming to meet sustainability goals. The process supports the commercial scale-up of complex dihydropyridines by offering a pathway that is both economically viable and environmentally responsible, ensuring long-term supply continuity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this amlodipine synthesis technology. These insights are derived directly from the experimental data and claims within patent CN102070516A, providing a factual basis for decision-making. Understanding these nuances helps R&D and procurement teams evaluate the feasibility of adopting this route for their specific manufacturing contexts.
Q: How does this novel synthesis method improve impurity profiles compared to traditional routes?
A: Traditional methods often require amino protection and subsequent deprotection using hydrazine or azanol, which generate difficult-to-remove Impurity A and Impurity B. This novel one-step nucleophilic substitution eliminates the need for protection groups entirely, thereby preventing the formation of these specific genotoxic or persistent impurities and ensuring higher final drug purity.
Q: What are the critical reaction parameters for scaling this amlodipine intermediate process?
A: The process relies on a nucleophilic substitution between a dihydropyridine derivative with a leaving group (such as chloro or mesylate) and aminoethanol. Critical parameters include the choice of base (e.g., sodium hydride, potassium tert-butoxide), the use of anhydrous aprotic solvents like THF or toluene to prevent side reactions, and maintaining temperatures between 0°C and 25°C to control exotherms and ensure selectivity.
Q: Can this manufacturing route support commercial-scale production volumes?
A: Yes, the route is designed for industrial applicability. It utilizes commercially available raw materials and avoids cryogenic conditions or hazardous deprotection reagents. The simplified workflow, involving fewer unit operations and standard workup procedures like extraction and crystallization, facilitates straightforward scale-up from pilot batches to multi-ton annual production capacities.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Amlodipine Supplier
The technological advancements detailed in CN102070516A represent a significant leap forward in the efficient production of cardiovascular therapeutics. At NINGBO INNO PHARMCHEM, we recognize the critical importance of translating such innovative laboratory methods into robust, industrial-scale realities. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facility is equipped with state-of-the-art reactors capable of handling the specific solvent and temperature requirements of this nucleophilic substitution process, ensuring that the theoretical benefits of high purity and yield are realized in every batch. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every gram of amlodipine intermediate we supply meets the highest global pharmacopoeial standards.
We invite global pharmaceutical partners to collaborate with us to leverage this advanced synthesis route for your supply chain. By partnering with our technical team, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you secure a sustainable, high-quality supply of amlodipine intermediates that drives value and efficiency in your drug development and manufacturing programs.