Revolutionizing Medetomidine Production: A High-Efficiency Three-Step Synthetic Route for Global Supply Chains
Revolutionizing Medetomidine Production: A High-Efficiency Three-Step Synthetic Route for Global Supply Chains
The pharmaceutical and veterinary industries are constantly seeking robust, scalable, and cost-effective methodologies for the synthesis of critical active pharmaceutical ingredients (APIs) and their intermediates. A significant breakthrough in this domain is detailed in patent CN103664788A, which discloses a novel method for preparing Medetomidine, a potent alpha-2 adrenoceptor agonist widely used for sedation and analgesia. This patent addresses the longstanding inefficiencies of conventional synthesis routes by introducing a streamlined three-step process that utilizes inexpensive raw materials such as 4-imidazole derivatives and 1-(2,3-dimethylphenyl)ethanone. By drastically reducing the number of reaction steps and eliminating the need for harsh cryogenic conditions or toxic heavy metal catalysts found in prior art, this technology offers a compelling value proposition for reliable pharmaceutical intermediate suppliers aiming to optimize their manufacturing portfolios. The method not only achieves a remarkable total yield of 76% but also ensures product purity levels reaching 99.5%, meeting the stringent quality standards required for global pharmacopoeias.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historical approaches to Medetomidine synthesis have been plagued by significant operational and economic drawbacks that hinder efficient commercial scale-up of complex pharmaceutical intermediates. For instance, earlier patents such as US4443466A describe routes involving Friedel-Crafts acylation with titanium tetrachloride, which often result in very low yields and generate substantial hazardous waste. Other methods, like those disclosed in J Chem Soc Perkin Trans, require extremely harsh reaction conditions, specifically maintaining temperatures at -78°C using butyl lithium, which imposes severe energy costs and requires specialized cryogenic infrastructure that is difficult to maintain in large-scale reactors. Furthermore, alternative routes disclosed in documents like CN102753532A involve up to seven reaction steps, including multiple Grignard reactions and high-pressure catalytic hydrogenation, while unfortunately utilizing contraindicated heavy metals like lead. These lengthy processes inevitably lead to cumulative yield losses, increased solvent consumption, and complex purification challenges, making them economically unviable for modern, high-volume manufacturing environments where cost reduction in pharmaceutical manufacturing is paramount.
The Novel Approach
In stark contrast to these cumbersome legacy methods, the technology outlined in CN103664788A presents a paradigm shift by condensing the synthesis into merely three highly efficient steps. This novel approach leverages cheap and readily available starting materials, specifically selecting 4-haloimidazoles and simple acetophenone derivatives, to construct the core scaffold of Medetomidine. The process cleverly utilizes an N-protection strategy followed by a single Grignard addition and a final one-pot reduction-deprotection sequence. This strategic simplification eliminates the need for repeated oxidation-reduction cycles and multiple protection-deprotection sequences that characterize older routes. By operating under mild conditions ranging from 0°C to 30°C for the initial steps and utilizing standard palladium-carbon catalysts for the final reduction, the method significantly lowers the barrier to entry for production. The result is a process that is not only easier to control and safer to operate but also delivers a high-purity product with minimal impurity profiles, directly addressing the critical needs of procurement managers focused on supply chain reliability and cost efficiency.
Mechanistic Insights into the Three-Step Catalytic Sequence
The core of this technological advancement lies in its elegant chemical logic, beginning with the strategic protection of the imidazole nitrogen to prevent unwanted side reactions during subsequent nucleophilic attacks. In the first step, a compound of formula 2, typically a 4-haloimidazole such as 4-iodo-imidazole, is contacted with a protecting agent of formula 3, such as N,N-dimethylsulfonyl chloride, in the presence of a base. This reaction effectively masks the acidic proton on the imidazole ring, generating a stable intermediate of formula 4. The choice of base, such as sodium hydroxide or triethylamine, and solvent, like tetrahydrofuran or acetonitrile, is critical to ensuring complete conversion without degrading the sensitive halogen substituent. This protection step is fundamental because it directs the subsequent Grignard reagent exclusively to the desired position, thereby enhancing the overall regioselectivity and purity of the final product.
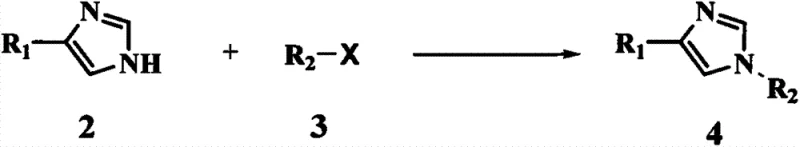
Following the protection phase, the synthesis proceeds to the crucial carbon-carbon bond-forming step, where the protected imidazole intermediate reacts with 1-(2,3-dimethylphenyl)ethanone. This transformation is achieved through a Grignard reaction using reagents such as isopropyl magnesium bromide, which acts as a powerful nucleophile to attack the carbonyl group of the ketone. The reaction is conducted in aprotic solvents like tetrahydrofuran at controlled temperatures between 15°C and 25°C, ensuring a smooth addition to form the tertiary alcohol intermediate of formula 6. This step is particularly notable for its efficiency; unlike prior art that might require multiple Grignard additions or oxidations, this single addition installs the necessary carbon framework in one go. The use of isopropyl magnesium bromide is preferred due to its balance of reactivity and availability, facilitating high conversion rates while minimizing the formation of homocoupling byproducts that could complicate downstream purification.

The final stage of the synthesis involves a sophisticated one-pot reduction and deprotection sequence that converts the alcohol intermediate directly into the target Medetomidine molecule. By subjecting the compound of formula 6 to catalytic hydrogenation in the presence of an acid, such as concentrated hydrochloric acid, and a palladium-carbon catalyst, two critical transformations occur simultaneously. The acid facilitates the cleavage of the N-protecting group (e.g., the sulfonyl group), while the hydrogen gas and catalyst reduce the benzylic alcohol functionality to the corresponding ethyl group. This tandem reaction is a masterstroke of process chemistry, as it combines what would traditionally be two separate unit operations into a single vessel operation. Conducted at moderate pressures of 0.1 to 0.6 MPa and temperatures of 65 to 85°C, this step not only saves time and energy but also reduces the exposure of the intermediate to potential degradation, thereby securing the high yield and 99.5% purity reported in the patent examples.

How to Synthesize Medetomidine Efficiently
The synthesis of Medetomidine via this patented route represents a significant optimization for process chemists aiming to establish a robust manufacturing protocol. The procedure begins with the dissolution of the halo-imidazole starting material in a suitable solvent, followed by the controlled addition of the protecting group reagent under basic conditions to ensure complete N-substitution. Once the protected intermediate is isolated and purified, typically through simple extraction and crystallization, it is subjected to the Grignard reaction with the specific acetophenone derivative. The final step involves loading the resulting alcohol intermediate into a hydrogenation reactor with acid and catalyst, followed by heating and pressurization to effect the simultaneous reduction and deprotection. For detailed standardized operating procedures, safety data, and specific stoichiometric ratios optimized for your specific reactor configuration, please refer to the technical guidelines below.
- Protect the imidazole nitrogen of a 4-haloimidazole derivative using a sulfonyl chloride or trityl chloride in the presence of a base to form the protected intermediate.
- React the protected imidazole with 1-(2,3-dimethylphenyl)ethanone using a Grignard reagent like isopropyl magnesium bromide to form the alcohol intermediate.
- Subject the alcohol intermediate to catalytic hydrogenation in the presence of an acid and palladium catalyst to simultaneously reduce and deprotect, yielding Medetomidine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this three-step synthesis route offers transformative benefits that extend far beyond simple chemical yield improvements. By collapsing a traditionally seven-step process into just three operations, the method drastically reduces the consumption of solvents, reagents, and energy, leading to substantial cost savings in pharmaceutical intermediate manufacturing. The elimination of cryogenic steps removes the need for expensive specialized equipment and the high energy costs associated with maintaining sub-zero temperatures, while the avoidance of toxic heavy metals like lead simplifies waste treatment and regulatory compliance. Furthermore, the use of cheap, commodity-grade starting materials ensures that the supply chain is less vulnerable to the volatility often seen with exotic or highly specialized reagents, thereby enhancing the overall stability and predictability of raw material sourcing for long-term production contracts.
- Cost Reduction in Manufacturing: The streamlined nature of this process directly translates to lower operational expenditures by minimizing the number of unit operations and isolation steps required. Each eliminated step in a synthetic route typically saves significant amounts of labor, solvent, and time, and in this case, the reduction from seven steps to three creates a compounding effect on efficiency. Additionally, the high atom economy of the Grignard addition and the one-pot final step means that less raw material is wasted as byproduct, further driving down the cost of goods sold (COGS) without compromising on the quality of the final API intermediate.
- Enhanced Supply Chain Reliability: Sourcing strategies are greatly improved by the reliance on widely available building blocks such as 4-iodo-imidazole and 2,3-dimethylacetophenone, which are produced by multiple global vendors. This diversification of supply sources mitigates the risk of single-supplier bottlenecks that can plague more complex syntheses relying on custom-synthesized precursors. Moreover, the robustness of the reaction conditions, which do not require extreme temperatures or pressures, ensures that production can be maintained consistently across different manufacturing sites, reducing the lead time for high-purity pharmaceutical intermediates and ensuring continuity of supply for downstream drug formulation.
- Scalability and Environmental Compliance: The process is inherently designed for industrial scale-up, utilizing standard reaction vessels and workup techniques like extraction and crystallization that are easily transferable from pilot plant to commercial tonnage. The absence of prohibited heavy metals and the reduction in hazardous waste generation align perfectly with modern green chemistry principles and strict environmental regulations. This compliance reduces the administrative burden and cost associated with waste disposal and environmental permitting, making the facility more sustainable and resilient against tightening global environmental standards while maintaining high throughput capabilities.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Medetomidine synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent literature, offering clarity on how this method outperforms historical precedents. Understanding these details is crucial for technical teams evaluating the feasibility of integrating this route into existing production lines or for procurement specialists assessing the long-term viability of the supply source.
Q: How does this new method improve yield compared to traditional Medetomidine synthesis?
A: Traditional methods often involve 7 or more steps with harsh conditions like -78°C or toxic heavy metals, leading to cumulative yield losses. This patented 3-step route achieves a total yield of approximately 76% with product purity reaching 99.5%, significantly minimizing material loss and purification burdens.
Q: What are the safety advantages of this synthetic route for industrial scale-up?
A: Unlike prior art requiring cryogenic temperatures (-78°C) or toxic lead catalysts, this process operates under mild conditions (0-30°C for initial steps) and utilizes standard catalytic hydrogenation. This eliminates the need for specialized cryogenic equipment and reduces environmental hazards associated with heavy metal waste disposal.
Q: Can this process be adapted for large-scale commercial production?
A: Yes, the process is specifically designed for industrial mass production. It uses cheap, readily available starting materials like 4-iodo-imidazole and 1-(2,3-dimethylphenyl)ethanone. The workup involves simple extraction and crystallization, avoiding complex chromatography, which makes it highly scalable from kilogram to multi-ton quantities.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Medetomidine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic methodologies to maintain competitiveness in the global pharmaceutical market. Our team of expert process chemists has thoroughly analyzed the technology disclosed in CN103664788A and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this efficient route to life. We are committed to delivering Medetomidine and its key intermediates with stringent purity specifications, leveraging our rigorous QC labs to ensure every batch meets the highest international standards. Our state-of-the-art facilities are equipped to handle the specific requirements of Grignard chemistry and catalytic hydrogenation safely and efficiently, ensuring a seamless transition from development to full-scale manufacturing.
We invite you to collaborate with us to leverage these process innovations for your supply chain. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out today to obtain specific COA data and route feasibility assessments that demonstrate how our optimized production capabilities can enhance your project's timeline and budget. Let us be your trusted partner in delivering high-quality chemical solutions that drive your business forward.