Advanced Solid-Phase Synthesis of Eptifibatide: A Scalable Route for High-Purity API Intermediates
Advanced Solid-Phase Synthesis of Eptifibatide: A Scalable Route for High-Purity API Intermediates
The pharmaceutical landscape for cardiovascular therapeutics is constantly evolving, driven by the need for more efficient and cost-effective manufacturing processes for critical antiplatelet agents. Patent CN101838308A discloses a robust solid-phase synthesis method for Eptifibatide, a cyclic heptapeptide that serves as a potent glycoprotein IIb/IIIa inhibitor. This technology represents a significant departure from traditional liquid-phase strategies, offering a streamlined pathway that addresses key bottlenecks in peptide manufacturing. The core innovation lies in the strategic selection of single-protected amino acid building blocks and the implementation of a rapid hydrogen peroxide oxidation step to form the critical disulfide bridge. By shifting from complex double-protection strategies to a simplified Fmoc-based solid-phase approach, this method not only enhances the accessibility of raw materials but also drastically reduces the production cycle time. For R&D directors and process chemists, this patent provides a blueprint for achieving high-purity pharmaceutical intermediates with improved operational simplicity. The structural integrity of the final molecule, characterized by the sequence Mpr-Har-Gly-Asp-Trp-Pro-Cys, is meticulously preserved through controlled cleavage and purification conditions.
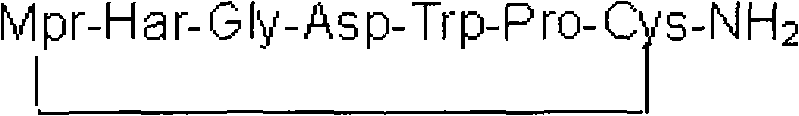
Furthermore, the transition to solid-phase synthesis allows for better control over impurity profiles, a critical factor for regulatory compliance in API production. The method described in CN101838308A eliminates the need for harsh environmental conditions associated with older BOC strategies, aligning with modern green chemistry principles. As a reliable API intermediate supplier, understanding these mechanistic nuances is essential for scaling production from laboratory grams to commercial tons. The ability to produce Eptifibatide acetate with consistent quality and reduced lead times positions this technology as a cornerstone for supply chain resilience in the cardiovascular drug sector.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the manufacturing of Eptifibatide has been dominated by liquid-phase synthesis methods, such as those employed by major legacy manufacturers like Bachem in Switzerland. These conventional routes typically utilize the BOC (tert-butyloxycarbonyl) protection strategy, which, while effective, presents significant logistical and environmental challenges. The liquid-phase synthesis cycle is inherently long, requiring multiple isolation and purification steps between each amino acid coupling, which accumulates time and material losses. Moreover, the BOC strategy often necessitates the use of hazardous reagents and generates substantial chemical waste, leading to higher environmental pollution burdens and increased disposal costs. Another critical bottleneck in traditional methods is the reliance on double-protected amino acids, such as Fmoc-Har(Pbf)-OH and Fmoc-Trp(Boc)-OH. These specialized building blocks are notoriously difficult to synthesize, often resulting in low yields and high market prices, which directly inflates the cost of goods sold (COGS). Additionally, the solubility of the linear peptide precursors in aqueous media is often poor, complicating the oxidation step required to form the cyclic structure and leading to inconsistent batch quality.
The Novel Approach
In stark contrast, the solid-phase synthesis method disclosed in patent CN101838308A introduces a paradigm shift by leveraging Fmoc chemistry on a Rink Amide AM Resin support. This approach fundamentally simplifies the synthetic route by utilizing single-protected amino acids, specifically Fmoc-Har-OH and Fmoc-Trp-OH, which are far more accessible and economical than their double-protected counterparts. The use of solid-phase support allows for the rapid washing away of excess reagents and by-products after each coupling step, driving reactions to completion and minimizing side reactions. A standout feature of this novel approach is the oxidation protocol; instead of relying on slow atmospheric oxidation or iodine-mediated methods that can take over 24 hours, this process employs hydrogen peroxide to achieve disulfide bond formation in merely 30 to 60 minutes. This dramatic reduction in reaction time translates directly to increased reactor throughput and lower energy consumption. Furthermore, the method incorporates a clever solubility enhancement step using acetonitrile and weak ammonia prior to oxidation, ensuring that the crude peptide remains in solution for efficient conversion. This combination of simplified raw materials, rapid kinetics, and improved solubility management makes the process highly attractive for cost reduction in pharmaceutical manufacturing.
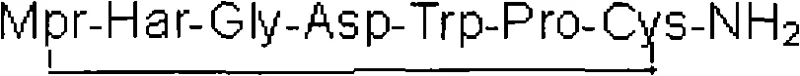
Mechanistic Insights into Fmoc-Based Solid-Phase Peptide Synthesis
The mechanistic elegance of this synthesis lies in the precise orchestration of amino acid condensation on the solid support. The process initiates with the loading of Fmoc-Cys(Trt)-OH onto the Fmoc-Rink-AM Resin, establishing the C-terminus of the peptide chain. Subsequent amino acids—Pro, Trp, Asp, Gly, Har, and finally Mpr—are coupled sequentially using standard activation reagents like HOBt and DIC. The choice of the Trt (trityl) protecting group for the cysteine thiol is crucial, as it remains stable during the repetitive piperidine deprotection cycles but is cleanly removed during the final acidic cleavage. The use of single-protected Fmoc-Har-OH avoids the steric hindrance and potential racemization issues often associated with bulky side-chain protecting groups like Pbf. Once the full linear sequence is assembled on the resin, the peptide is cleaved using a high concentration of trifluoroacetic acid (87.5% TFA) at controlled temperatures (around 30°C). This step simultaneously removes the side-chain protecting groups and releases the linear peptide into the solution. The resulting linear precursor, Mpr-Har-Gly-Asp-Trp-Pro-Cys-NH2, contains free thiol groups on the Mpr and Cys residues that are primed for oxidation.
The formation of the intramolecular disulfide bond is the critical cyclization step that confers biological activity to Eptifibatide. In this patented method, the linear crude product is first dissolved in water, potentially aided by acetonitrile to overcome solubility limits. The pH is adjusted to neutrality using weak ammonia, creating an optimal environment for the subsequent oxidation. Hydrogen peroxide is then introduced at a concentration of 0.1% to 0.5% of the crude product volume. The mechanism involves the oxidation of the two free sulfhydryl (-SH) groups to form a covalent disulfide (-S-S-) bridge, closing the peptide ring. This reaction is monitored via HPLC or MS to ensure completion within the 30 to 60-minute window. Following oxidation, the mixture is neutralized with acetic acid, filtered to remove any particulates, and subjected to preparative HPLC purification. The collection of characteristic peaks ensures the isolation of the target cyclic peptide from any linear by-products or deletion sequences. Finally, the purified fractions are concentrated and freeze-dried to yield the final Eptifibatide acetate salt, as depicted in the structural representation below.
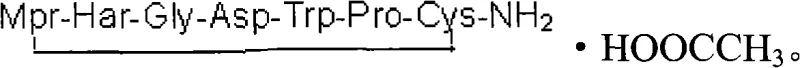
How to Synthesize Eptifibatide Efficiently
The synthesis of Eptifibatide via this solid-phase route requires careful attention to resin swelling, coupling efficiency, and oxidation conditions to ensure high purity and yield. The process is designed to be operationally simple, allowing for scalability without compromising on the stringent quality standards required for cardiovascular APIs. Below is a summary of the critical operational phases involved in transforming raw amino acids into the refined acetate salt. For detailed standard operating procedures and specific molar ratios, please refer to the technical guide below.
- Perform sequential amino acid condensation on Fmoc-Rink Amide Resin using single-protected Fmoc-amino acids (Cys, Pro, Trp, Asp, Gly, Har) and Mpr.
- Cleave the linear peptide from the resin using 87.5% trifluoroacetic acid (TFA) and precipitate with ice ether to obtain the crude linear product.
- Oxidize the dissolved crude product with 0.1-0.5% hydrogen peroxide for 30-60 minutes to form the disulfide bond, followed by HPLC purification and freeze-drying.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of the synthesis method described in CN101838308A offers tangible strategic benefits that extend beyond mere technical feasibility. The primary advantage lies in the radical simplification of the raw material supply chain. By eliminating the dependency on scarce and expensive double-protected amino acids, manufacturers can source standard Fmoc-amino acids from a broader range of global suppliers, thereby reducing supply risk and negotiating leverage. This shift significantly lowers the barrier to entry for production and stabilizes the cost base against fluctuations in specialty chemical markets. Moreover, the drastic reduction in reaction time for the oxidation step—from days to less than an hour—means that manufacturing assets are utilized more efficiently, allowing for higher production volumes within the same timeframe. This increased throughput capability is essential for meeting sudden spikes in demand for generic cardiovascular medications without the need for massive capital expenditure on new facilities.
- Cost Reduction in Manufacturing: The economic impact of switching to single-protected amino acids cannot be overstated. Double-protected variants like Fmoc-Har(Pbf)-OH involve complex multi-step syntheses themselves, commanding premium prices and long lead times. By utilizing Fmoc-Har-OH and Fmoc-Trp-OH, the direct material cost is substantially decreased. Additionally, the use of hydrogen peroxide as an oxidant is far more cost-effective than using stoichiometric amounts of iodine or relying on slow air oxidation which ties up reactor capacity. The elimination of expensive heavy metal catalysts or complex purification steps further drives down the overall processing cost, resulting in a more competitive price point for the final API intermediate.
- Enhanced Supply Chain Reliability: Supply chain continuity is often threatened by the availability of niche reagents. The conventional reliance on specific double-protected building blocks creates a single point of failure in the supply chain. In contrast, the single-protected amino acids required for this novel route are commodity chemicals within the peptide synthesis industry, available from multiple qualified vendors worldwide. This diversification of the supplier base ensures that production schedules are not disrupted by shortages of a single specialized ingredient. Furthermore, the shortened production cycle means that inventory turnover is faster, allowing manufacturers to respond more agilely to market demands and reduce the working capital tied up in work-in-progress inventory.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, solid-phase synthesis is inherently more amenable to automation and scale-up than liquid-phase methods. The waste streams generated are more predictable and easier to manage, particularly when avoiding the heavy metal waste associated with some traditional oxidation methods. The use of hydrogen peroxide, which decomposes into water and oxygen, aligns with green chemistry initiatives and reduces the burden on wastewater treatment facilities. This environmental compatibility simplifies the regulatory approval process for new manufacturing sites and ensures long-term operational sustainability. The robustness of the process allows for seamless scaling from pilot batches to multi-ton commercial production, ensuring a steady supply of high-purity Eptifibatide for the global market.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the solid-phase synthesis of Eptifibatide. These insights are derived directly from the experimental data and advantageous effects reported in patent CN101838308A, providing clarity on process optimization and quality control measures. Understanding these details is crucial for technical teams evaluating the feasibility of technology transfer.
Q: Why is hydrogen peroxide oxidation preferred over traditional methods for Eptifibatide synthesis?
A: Traditional atmospheric or iodine oxidation methods often require over 24 hours to complete the disulfide bond formation. The patented method utilizes hydrogen peroxide oxidation, which drastically reduces the reaction time to just 30-60 minutes, significantly shortening the overall production cycle and improving throughput efficiency.
Q: What are the advantages of using single-protected amino acids in this route?
A: Conventional routes often rely on difficult-to-synthesize double-protected amino acids like Fmoc-Har(Pbf)-OH or Fmoc-Trp(Boc)-OH, which are expensive and hard to source with high purity. This novel approach utilizes single-protected variants (Fmoc-Har-OH and Fmoc-Trp-OH), which are more economically practical, easier to obtain commercially, and help improve the purity of the crude peptide product.
Q: How does this process address the solubility issues of the crude peptide?
A: The linear crude product of Eptifibatide often exhibits poor water solubility, complicating purification. This method solves the issue by first adding an organic solvent like acetonitrile to enhance solubility, adjusting the pH to neutrality with weak ammonia, and then proceeding with oxidation. This ensures a homogeneous reaction environment and facilitates subsequent HPLC purification.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Eptifibatide Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust synthetic routes in the production of life-saving cardiovascular medications. Our team of expert process chemists has extensively analyzed the solid-phase synthesis technology disclosed in CN101838308A and is fully equipped to implement this advanced methodology at commercial scale. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with unwavering consistency. Our state-of-the-art facilities are designed to handle complex peptide synthesis with stringent purity specifications, utilizing rigorous QC labs to verify every batch against the highest international standards. We understand that the transition from lab-scale innovation to industrial reality requires not just equipment, but deep technical expertise in managing resin handling, cleavage kinetics, and purification chromatography.
We invite you to collaborate with us to leverage this cost-effective and efficient synthesis route for your Eptifibatide requirements. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume needs, demonstrating how this single-protected amino acid strategy can optimize your budget. Please contact us today to request specific COA data and route feasibility assessments. By partnering with NINGBO INNO PHARMCHEM, you secure a supply chain that is not only reliable and compliant but also optimized for the future of pharmaceutical manufacturing.