Advanced Synthesis of Alpha-Acyloxy Thioether Derivatives for Pharmaceutical Manufacturing
The landscape of fine chemical synthesis is constantly evolving, driven by the need for greener, more efficient pathways to complex organic scaffolds. A significant breakthrough in this domain is detailed in patent CN112898188B, which discloses a novel method for preparing alpha-acyloxy thioether derivatives. These compounds serve as vital building blocks in organic synthesis, particularly for constructing aldehyde and ketone functionalities found in various natural products and pharmaceutical agents. The disclosed technology represents a paradigm shift from traditional, hazardous routes to a streamlined, catalytic approach that leverages the unique reactivity of electrophilic fluorinating agents. By utilizing a combination of simple carboxylic acids, their corresponding sodium salts, and the commercial reagent Selectfluor, this method achieves high yields under relatively mild conditions. For R&D directors and process chemists, this innovation offers a robust solution to the long-standing challenges associated with sulfur functionalization, promising enhanced purity profiles and simplified downstream processing. As a reliable pharmaceutical intermediate supplier, understanding such cutting-edge methodologies is crucial for maintaining a competitive edge in the global market.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of alpha-acyloxy thioethers has been plagued by significant operational and safety hurdles that hinder large-scale adoption. The most prevalent traditional strategy involves the Pummerer rearrangement, which necessitates the prior preparation of sulfoxide intermediates. This prerequisite not only adds extra synthetic steps, thereby increasing overall production time and cost, but also introduces substantial toxicity concerns associated with handling sulfoxide compounds. Furthermore, alternative methods relying on thioether substrates often demand the use of expensive transition metal catalysts or high-valence iodine reagents. These reagents are not only costly but can exhibit unpredictable reactivity profiles, leading to inconsistent yields and difficult-to-remove metal impurities that compromise the quality of the final API intermediate. The reliance on such harsh or expensive reagents creates a bottleneck for cost reduction in pharmaceutical intermediate manufacturing, forcing companies to absorb higher raw material costs and invest in complex purification infrastructure to meet stringent regulatory standards for residual metals.
The Novel Approach
In stark contrast to these legacy methods, the technology described in patent CN112898188B introduces a direct, one-pot functionalization strategy that bypasses the need for pre-oxidized sulfoxides. The core of this innovation lies in the use of Selectfluor (1-chloromethyl-4-fluoro-1,4-diazabicyclo[2.2.2]octane bis(tetrafluoroborate)) as a potent electrophilic fluorinating agent. This reagent activates the thioether substrate in situ, enabling a cascade reaction that installs the acyloxy group with high regioselectivity. The reaction scheme below illustrates the general transformation, where an N-substituted-2-methylthiobenzamide reacts with a carboxylic acid source to yield the target alpha-acyloxy thioether derivative efficiently.
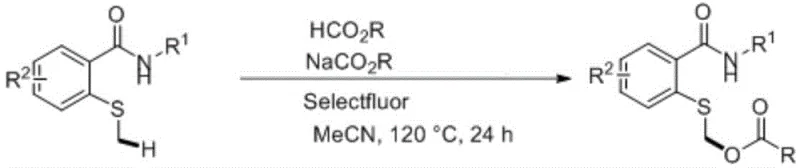
This approach drastically simplifies the synthetic route by merging oxidation and functionalization into a single operational step. By eliminating the need for toxic sulfoxide precursors and expensive metal catalysts, the process aligns perfectly with green chemistry principles. The use of readily available carboxylic acids and their salts as nucleophilic partners further enhances the economic viability of the process. For procurement teams, this translates to a supply chain less dependent on volatile specialty reagent markets. The method's ability to tolerate various substituents on the aromatic ring and the amide nitrogen demonstrates its versatility, making it a powerful tool for generating diverse libraries of high-purity pharmaceutical intermediates required for drug discovery and development pipelines.
Mechanistic Insights into Selectfluor-Mediated C-H Functionalization
The mechanistic pathway of this transformation is a fascinating example of electrophilic activation followed by intramolecular rearrangement. The reaction initiates when the sulfur atom of the thioether substrate attacks the electrophilic fluorine of the Selectfluor reagent, generating a reactive fluorosulfonium ion intermediate. This species is highly electrophilic and susceptible to nucleophilic attack. In the presence of the carboxylate anion (derived from the sodium salt of the carboxylic acid), an intramolecular cyclization occurs, forming a cyclic cationic intermediate. This step is critical as it locks the conformation and directs the subsequent bond-breaking events. The cyclic intermediate then undergoes elimination of a proton at the alpha-position relative to the sulfur, facilitated by the basic environment provided by the acetate salt. This elimination triggers the cleavage of the nitrogen-sulfur bond, releasing a sulfur-centered cation and an isomeric product. Finally, the nucleophilic carboxylate anion attacks the electrophilic carbon adjacent to the sulfur, yielding the stable alpha-acyloxy thioether product. This intricate dance of bond formation and cleavage ensures high regioselectivity, minimizing the formation of unwanted byproducts.
Impurity control is inherently built into this mechanism through the requirement of the ortho-amide directing group. Comparative experiments within the patent data reveal that substrates lacking the ortho-amide functionality or possessing N,N-dialkyl substitution fail to yield the desired product. This suggests that the amide hydrogen plays a pivotal role, likely through hydrogen bonding or coordination that stabilizes the transition state or the fluorosulfonium intermediate. This intrinsic selectivity acts as a chemical filter, preventing the reaction from proceeding on non-compliant substrates and thus simplifying the impurity profile of the crude reaction mixture. For quality control laboratories, this means fewer unknown peaks in HPLC chromatograms and a more straightforward path to meeting specification limits. Understanding these mechanistic nuances allows process chemists to fine-tune reaction parameters, such as temperature and stoichiometry, to maximize conversion while suppressing potential side reactions like over-oxidation or polymerization.
How to Synthesize Alpha-Acyloxy Thioether Derivatives Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires careful attention to reaction conditions to ensure optimal yield and safety. The general procedure involves charging a sealed, pressure-resistant tube with the thioether substrate, the chosen carboxylic acid, its sodium salt, and Selectfluor in acetonitrile solvent. The reaction mixture is then heated to temperatures ranging from 100 °C to 150 °C, with 120 °C identified as the optimal setpoint for balancing reaction rate and safety. A typical reaction time spans 24 hours, allowing sufficient time for the complete consumption of the starting material. The specific example below demonstrates the successful synthesis of methyl [(2-n-butylcarbamoylphenyl)thio]acetate, achieving an impressive yield of 87% under these standardized conditions.
- Charge a sealed pressure-resistant tube with acetonitrile solvent, N-substituted-2-methylthiobenzamide substrate, carboxylic acid, sodium carboxylate salt, and Selectfluor reagent.
- Heat the reaction mixture to a temperature between 100-150 °C, preferably 120 °C, and stir vigorously for 12 to 36 hours, optimally 24 hours.
- Upon completion, distill off the acetonitrile solvent and purify the resulting crude residue via column chromatography using dichloromethane as the eluent.

Following the reaction period, the workup procedure is remarkably straightforward, reflecting the process's efficiency. The reaction solvent, acetonitrile, is removed via distillation, leaving behind a crude residue containing the product and inorganic salts. This residue is then subjected to column chromatography, typically using dichloromethane as the eluent, to isolate the pure alpha-acyloxy thioether derivative. The simplicity of this isolation step is a major advantage over metal-catalyzed processes that often require tedious scavenging steps to remove trace metals. For detailed operational parameters and specific stoichiometric ratios tailored to different substrates, please refer to the standardized protocol below.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this Selectfluor-mediated methodology offers profound advantages that extend beyond mere chemical elegance. For procurement managers tasked with optimizing raw material spend, the elimination of expensive hypervalent iodine reagents and transition metal catalysts represents a significant opportunity for cost reduction in fine chemical manufacturing. The reagents employed—carboxylic acids, sodium salts, and Selectfluor—are commodity chemicals available from multiple global suppliers, mitigating the risk of supply chain disruptions associated with sole-source specialty catalysts. Furthermore, the high yields reported in the patent examples, often exceeding 70-80%, mean that less raw material is wasted per kilogram of product produced, directly improving the cost of goods sold (COGS). The simplified workup procedure also reduces the consumption of solvents and silica gel during purification, contributing to lower operational expenditures.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven by the replacement of costly and hazardous reagents with inexpensive, commercially available alternatives. By avoiding the use of transition metals, manufacturers eliminate the need for expensive metal scavengers and the associated analytical testing for residual metals, which is a mandatory and costly step in API production. Additionally, the high atom economy and yield of the reaction minimize waste disposal costs. The process operates in a common solvent like acetonitrile, which is easily recovered and recycled, further enhancing the overall economic efficiency of the production line. These factors combine to create a leaner, more cost-effective manufacturing process that improves margin potential for high-volume intermediates.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the use of robust, non-proprietary reagents. Unlike specialized ligands or catalysts that may have long lead times or limited production capacity, the key components of this reaction are produced on a multi-ton scale globally. This abundance ensures that production schedules are not held hostage by raw material shortages. Moreover, the reaction conditions are relatively forgiving, with a broad temperature window (100-150 °C) that accommodates standard heating equipment found in most multipurpose chemical plants. This flexibility allows for seamless technology transfer between different manufacturing sites, ensuring continuity of supply even if one facility faces maintenance or regulatory issues. The stability of the reagents also simplifies storage and logistics, reducing the risk of degradation during transport.
- Scalability and Environmental Compliance: Scaling this process from gram to ton scale is facilitated by its homogeneous nature and lack of exothermic hazards associated with strong oxidants like peracids. The use of sealed tubes in the patent examples translates well to autoclaves or pressurized reactors in a plant setting. From an environmental standpoint, the process adheres to green chemistry principles by avoiding toxic sulfoxides and heavy metals. This results in a cleaner waste stream that is easier and cheaper to treat, helping manufacturers meet increasingly stringent environmental regulations. The reduction in hazardous waste generation not only lowers disposal fees but also enhances the company's sustainability profile, a key metric for modern pharmaceutical clients who prioritize eco-friendly supply chains.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating this technology for their specific applications, we have compiled answers to common questions regarding the scope and limitations of the method. These insights are derived directly from the experimental data and mechanistic studies presented in the patent literature. Understanding these nuances is essential for successful process implementation and risk mitigation.
Q: What is the critical role of the ortho-amide group in this synthesis?
A: The ortho-amide group acts as a crucial directing group. Comparative studies indicate that substrates lacking this group or having N,N-dialkyl substitution fail to react, suggesting the amide hydrogen and proximity are essential for stabilizing the fluoronium intermediate and facilitating the subsequent intramolecular cyclization.
Q: Why is Selectfluor preferred over traditional hypervalent iodine reagents?
A: Selectfluor offers a safer, more cost-effective, and operationally simpler alternative to expensive and highly active hypervalent iodine reagents. It eliminates the need for toxic sulfoxide precursors required in Pummerer rearrangements, aligning with green chemistry principles and reducing hazardous waste generation.
Q: Can this method be scaled for industrial production?
A: Yes, the process utilizes commercially available reagents and standard solvents like acetonitrile. The reaction conditions (120 °C in sealed tubes) are manageable, and the workup involves simple solvent removal followed by chromatography, making it highly suitable for scale-up from laboratory to commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Alpha-Acyloxy Thioether Derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced synthetic methodologies like the Selectfluor-mediated functionalization of thioethers. Our team of expert process chemists is dedicated to translating such innovative academic and patent technologies into robust, commercial-scale manufacturing processes. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from lab bench to pilot plant is seamless and efficient. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of alpha-acyloxy thioether derivatives meets the exacting standards required by the global pharmaceutical industry. We understand that consistency and quality are non-negotiable in API synthesis, and our commitment to excellence ensures a reliable supply of critical intermediates.
We invite you to collaborate with us to leverage this cutting-edge chemistry for your next project. Whether you require custom synthesis of novel derivatives or scale-up of existing routes, our technical sales team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact our technical procurement team today to request specific COA data and discuss route feasibility assessments. By partnering with NINGBO INNO PHARMCHEM, you gain access to a supply chain partner that combines technical expertise with commercial agility, driving value and innovation in your drug development pipeline.