Optimizing Tenofovir Disoproxil Fumarate Production for Global Pharmaceutical Supply Chains
Introduction to Advanced TDF Manufacturing Technologies
The global demand for antiretroviral therapies continues to drive the need for robust, high-yield synthetic routes for critical active pharmaceutical ingredients (APIs). Patent CN103880884A introduces a refined methodology for the preparation of high-purity Tenofovir Disoproxil Fumarate (TDF), a cornerstone prodrug in the treatment of HIV and Hepatitis B. This technology addresses the persistent challenges of impurity control and yield optimization that have plagued earlier generations of synthesis protocols. By implementing a sophisticated acid-base extraction strategy, the process effectively isolates the target molecule from structurally similar byproducts, ensuring a final product purity that consistently exceeds regulatory thresholds. 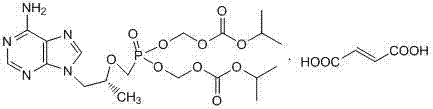
The significance of this innovation lies in its ability to transform a crude reaction mixture into a pharmaceutical-grade intermediate without resorting to costly and time-consuming chromatographic separations. For R&D directors and process chemists, understanding the nuances of this purification cycle is essential for replicating the high quality standards required by major health authorities. The method not only enhances the chemical integrity of the final API but also streamlines the downstream processing steps, offering a compelling value proposition for manufacturers seeking to optimize their production lines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of TDF has relied on straightforward condensation reactions followed by basic aqueous workups, as seen in earlier patents like US5922695. While these methods are conceptually simple, they suffer from significant drawbacks regarding impurity clearance. Specifically, standard washing procedures often fail to remove acidic impurities and unreacted starting materials effectively. A particularly stubborn contaminant is Tenofovir Monoisopropoxycarbonyloxymethyl Ester, a structural analog that co-crystallizes with the desired product. 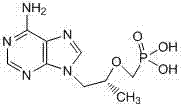
Furthermore, traditional crystallization techniques used to purify the free base often result in substantial product loss, dragging down overall molar yields. The inability to selectively partition the product away from these closely related impurities means that multiple recrystallization steps are often required, increasing solvent consumption and processing time. This inefficiency creates a bottleneck in the supply chain, limiting the ability to produce large quantities of high-purity material cost-effectively.
The Novel Approach
The methodology disclosed in CN103880884A breaks this cycle by introducing a targeted pH-controlled extraction sequence. Instead of a single wash, the process employs a dual-phase separation where the organic reaction mixture is first acidified to a specific pH range of 1.0 to 3.0. This step protonates the basic adenine moiety of the TDF free base, forcing it into the aqueous layer while leaving neutral organic impurities, such as excess chloromethyl isopropyl carbonate, behind in the organic solvent. Subsequently, the aqueous layer is basified to a pH of 7.0 to 9.5, regenerating the free base which is then extracted back into a fresh organic solvent. This 'acid-base swing' acts as a powerful purification filter, removing the majority of acidic and neutral contaminants in a single operational sequence.
Mechanistic Insights into Condensation and pH-Swing Extraction
The core chemical transformation involves the nucleophilic substitution of the phosphonate hydroxyl group of Tenofovir with chloromethyl isopropyl carbonate. Catalyzed by triethylamine in polar aprotic solvents like N-Methyl pyrrolidone (NMP) or DMF, this reaction proceeds efficiently at temperatures between 50°C and 70°C. 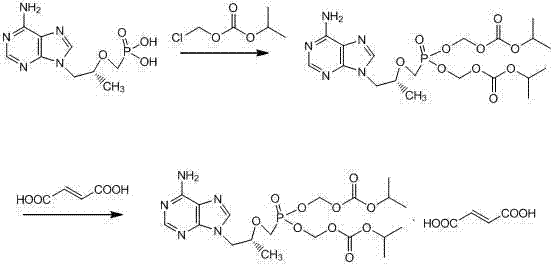
However, the true mechanistic brilliance of this patent lies in the workup phase. The differential solubility of the protonated versus neutral forms of the molecule is exploited to achieve high purity. When the pH is lowered, the product becomes a water-soluble salt, effectively separating it from lipophilic side products. Upon raising the pH, the product reverts to its lipophilic free base form. This mechanism ensures that the tenofovir monoester impurity, which lacks the second carbonate group and exhibits different solubility characteristics, is largely excluded from the final organic extract. Consequently, the resulting oil possesses a purity greater than 85% prior to salt formation, allowing the final fumarate salt crystallization to easily achieve purities above 99%.
How to Synthesize Tenofovir Disoproxil Fumarate Efficiently
To implement this high-yield synthesis, operators must strictly adhere to the temperature controls and pH adjustments detailed in the patent examples. The process begins with the condensation of Tenofovir and chloromethyl isopropyl carbonate, followed by the critical purification steps that define the quality of the output. The detailed standardized synthesis steps are provided in the guide below.
- Condense Tenofovir with chloromethyl isopropyl carbonate in polar aprotic solvents like NMP or DMF using triethylamine.
- Perform acid-base extraction: Acidify organic layer to pH 1.0-3.0 to transfer product to aqueous phase, removing neutral impurities.
- Basify aqueous phase to pH 7.0-9.5 to recover free base into organic solvent, followed by fumarate salt formation.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthesis route offers tangible benefits in terms of cost structure and reliability. By eliminating the need for extensive recrystallization or column chromatography, the process significantly reduces solvent usage and processing time. The simplified workflow translates directly into lower operational expenditures and a reduced environmental footprint, aligning with modern green chemistry initiatives.
- Cost Reduction in Manufacturing: The enhanced purification efficiency means that less raw material is wasted during the cleaning process. By achieving high purity early in the workup stage, manufacturers can avoid the yield losses associated with repetitive crystallization cycles. This logical reduction in processing steps leads to substantial cost savings in utility consumption and labor, making the production of high-purity TDF more economically viable.
- Enhanced Supply Chain Reliability: The robustness of the acid-base extraction method ensures consistent batch-to-batch quality. Unlike methods sensitive to minor fluctuations in cooling rates or seeding, this liquid-liquid extraction approach is highly reproducible. This consistency minimizes the risk of batch failures and rejections, thereby securing a steady flow of material for downstream formulation and reducing lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The use of common industrial solvents and standard extraction equipment facilitates easy scale-up from pilot to commercial production. Furthermore, the effective removal of impurities reduces the burden on waste treatment systems, as the effluent streams are cleaner and easier to manage. This scalability supports the commercial scale-up of complex nucleotide analogs without requiring specialized or exotic infrastructure.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and benefits of this specific TDF synthesis protocol. These insights are derived directly from the experimental data and claims presented in the patent documentation.
Q: How does this method improve impurity profiles compared to standard washing?
A: Unlike conventional water washing which leaves acidic impurities behind, this method utilizes a pH-swing extraction. By acidifying the organic layer, the basic product moves to the aqueous phase while neutral impurities like excess chloromethyl isopropyl carbonate remain in the organic layer, significantly reducing the tenofovir monoester content to below 0.5%.
Q: What solvents are compatible with this high-purity synthesis route?
A: The patent specifies polar aprotic solvents such as N-Methyl pyrrolidone (NMP), Dimethylformamide (DMF), or Dimethyl sulfoxide (DMSO) for the initial condensation reaction, ensuring high solubility and reaction efficiency at temperatures between 50°C and 70°C.
Q: Is this process suitable for large-scale industrial manufacturing?
A: Yes, the process avoids complex chromatographic purification, relying instead on scalable liquid-liquid extraction and crystallization. The reported molar yields exceed 65%, and the operational simplicity makes it highly adaptable for commercial scale-up of complex pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tenofovir Disoproxil Fumarate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of purity and yield in the production of antiviral APIs. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated extraction techniques described in CN103880884A can be seamlessly integrated into your supply chain. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of Tenofovir Disoproxil Fumarate meets the highest international standards.
We invite you to collaborate with us to leverage these advanced manufacturing capabilities for your next project. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our optimized processes can enhance your product portfolio.