Optimizing Tenofovir Impurity Standards: A Strategic Breakthrough in Pharmaceutical Intermediate Manufacturing
Optimizing Tenofovir Impurity Standards: A Strategic Breakthrough in Pharmaceutical Intermediate Manufacturing
The escalating demand for high-purity antiviral medications has placed intense scrutiny on the quality control of Active Pharmaceutical Ingredients (APIs), particularly for complex nucleotide analogs like Tenofovir Alafenamide (TAF). As regulatory bodies enforce stricter limits on degradation products, the ability to synthesize authentic impurity standards becomes a critical bottleneck in the pharmaceutical supply chain. Patent CN113185552A introduces a transformative preparation method for a specific Tenofovir Disoproxil Fumarate degradation impurity, known as Compound I, addressing the longstanding challenges of selectivity and purification in this niche sector. This technical disclosure outlines a robust two-phase hydrolysis strategy that leverages lithium hydroxide to achieve unprecedented purity levels exceeding 98%, offering a viable pathway for reliable pharmaceutical intermediates supplier networks to secure their quality assurance protocols.
The significance of this innovation extends beyond mere academic interest; it directly impacts the commercial viability of generic TAF production by enabling precise quantitative analysis of degradation pathways. By establishing a reliable source for Compound I, manufacturers can better monitor the stability of their final drug products, ensuring patient safety and regulatory compliance. The method described circumvents the pitfalls of previous synthetic routes, which often suffered from low yields and cumbersome isolation procedures. For R&D directors and procurement specialists alike, understanding the mechanistic advantages of this new protocol is essential for optimizing cost structures and ensuring the continuity of supply for these vital reference materials in the competitive antiviral market.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Tenofovir degradation impurities has relied heavily on acidic hydrolysis conditions, typically utilizing strong acids like trifluoroacetic acid to cleave specific ester bonds. As illustrated in the prior art reaction schemes, this approach involves converting a precursor impurity (Compound II) into the target Compound I under harsh acidic environments. However, this conventional methodology is fraught with significant technical drawbacks that hinder its scalability and economic efficiency. The primary issue lies in the lack of chemoselectivity; the aggressive acidic conditions often lead to continuous hydrolysis of the target product itself, resulting in a complex mixture of over-degraded by-products that are difficult to separate.
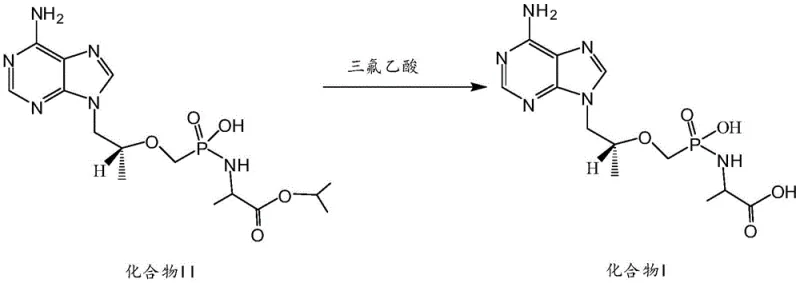
Furthermore, the use of trifluoroacetic acid introduces severe post-treatment complications. The target Compound I tends to form stable salts with the excess acid, necessitating elaborate neutralization and extraction steps that drastically reduce overall yield. From a supply chain perspective, the starting material, Compound II, is not a readily available commodity chemical, creating a dependency on specialized suppliers and inflating raw material costs. The combination of low selectivity, difficult purification, and scarce starting materials renders this traditional route unsuitable for the large-scale production of high-purity impurity standards required by modern pharmaceutical quality control laboratories.
The Novel Approach
In stark contrast to the acidic degradation pathways, the novel method disclosed in the patent utilizes a mild, selective hydrolysis driven by an alkaline two-phase system. This innovative route starts directly from Tenofovir Disoproxil free base, a much more accessible and cost-effective starting material compared to the specialized precursors required by older methods. The reaction employs an aqueous solution of lithium hydroxide in conjunction with a chlorine-containing organic solvent, such as dichloromethane, to create a controlled interfacial environment. This setup allows for the precise cleavage of the isopropyl alanine ester bond while leaving the critical phosphonate and phosphate linkages intact, a feat that acidic methods struggle to achieve.
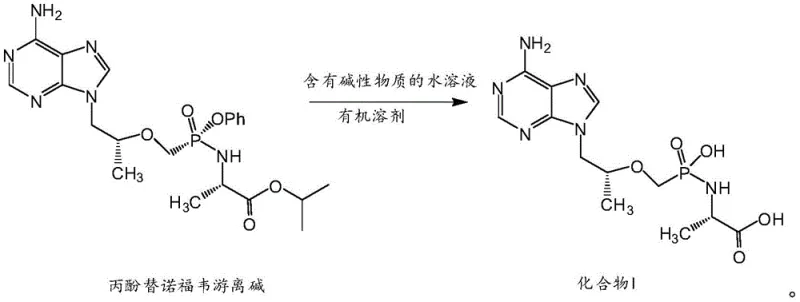
The operational simplicity of this new approach is a game-changer for industrial application. Post-reaction processing is streamlined; once the hydrolysis is complete, the reaction mixture naturally separates into organic and aqueous layers. The target product resides in the aqueous phase, allowing for the easy removal of organic impurities and unreacted starting materials simply by discarding the organic layer. Subsequent pH adjustment and purification via medium-pressure preparative chromatography yield the final product with exceptional purity. This shift from a complex, salt-forming acidic process to a clean, phase-separating alkaline process represents a substantial advancement in process chemistry, offering significant reductions in processing time and waste generation.
Mechanistic Insights into Selective Alkaline Hydrolysis
To fully appreciate the technical superiority of this method, one must delve into the molecular architecture of the Tenofovir Disoproxil free base and its susceptibility to hydrolytic attack. The molecule possesses multiple potential reactive centers, often referred to as active sites, which vary in their sensitivity to pH and solvent polarity. As detailed in the structural analysis, there are three distinct active sites vulnerable to hydrolysis: the phenyl phosphate ester bond (Active Site 1), the alanine carboxyl ester bond (Active Site 2), and the phosphonate linkage (Active Site 3). The challenge for any synthetic chemist is to trigger hydrolysis exclusively at Active Site 2 to generate Compound I, without inadvertently cleaving the phosphate ester or the phosphonate bond, which would lead to useless degradation products.
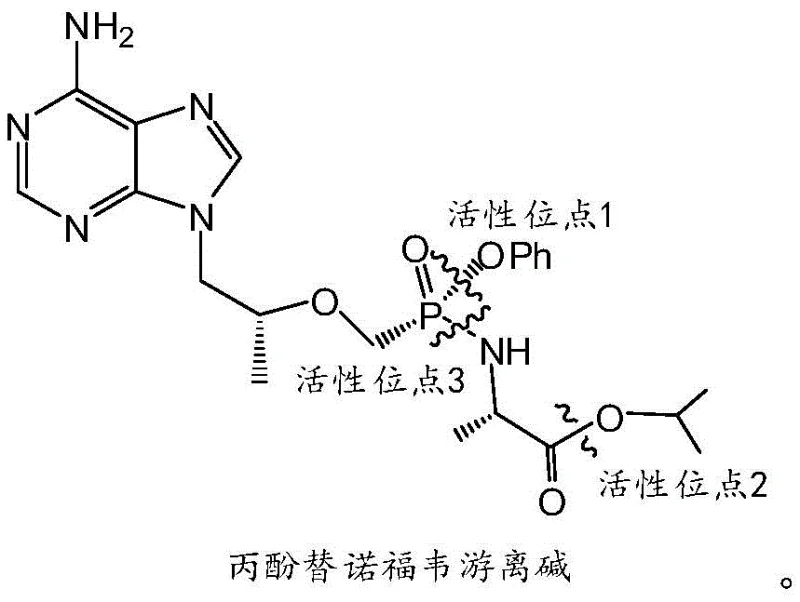
The selection of lithium hydroxide as the base is not arbitrary but is rooted in its unique physicochemical properties within a biphasic system. Unlike stronger alkali metal hydroxides such as potassium or sodium hydroxide, which were shown in comparative examples to cause excessive hydrolysis or no reaction at all depending on conditions, lithium hydroxide offers a "Goldilocks" level of reactivity. In the presence of the organic solvent, the lithium cation likely coordinates with the oxygen atoms of the ester group, activating it for nucleophilic attack by the hydroxide ion specifically at the alanine ester position. The two-phase nature of the reaction further modulates this reactivity, effectively shielding the more sensitive phosphate bonds from the aqueous base until the desired transformation is complete. This delicate balance ensures that the reaction stops at Compound I, preventing the formation of the fully de-esterified Tenofovir parent drug or other fragmented species.
Moreover, the control of reaction parameters such as temperature and molar ratios plays a pivotal role in directing the reaction pathway. The patent data indicates that maintaining the temperature between 10°C and 40°C, and specifically optimizing the molar ratio of free base to lithium hydroxide around 1:4, maximizes the conversion to the desired impurity. Deviations from these optimal conditions result in either incomplete reaction or the onset of side reactions at the other active sites. This mechanistic understanding underscores the importance of precise process control in the manufacturing of high-purity pharmaceutical intermediates, where minor deviations can lead to significant losses in yield and purity.
How to Synthesize Tenofovir Disoproxil Fumarate Degradation Impurity Efficiently
Implementing this synthesis protocol requires strict adherence to the optimized reaction conditions to ensure reproducibility and high purity. The process is designed to be scalable, moving seamlessly from laboratory benchtop to pilot plant operations with minimal modification. The key to success lies in the preparation of the two-phase system and the careful monitoring of the hydrolysis progress. Operators must ensure that the lithium hydroxide concentration is within the specified range of 2.0 to 5.0 mol/L to maintain the necessary reaction kinetics without overwhelming the system. The use of medium-pressure preparative chromatography in the final purification step is critical for removing trace impurities and achieving the >98% purity required for reference standards.
- Mix Tenofovir Disoproxil free base with lithium hydroxide aqueous solution and a chlorine-containing organic solvent to initiate a controlled two-phase reaction.
- Maintain the reaction temperature between 10°C and 40°C for 10 to 20 hours to ensure selective hydrolysis without over-degradation.
- Separate the layers, adjust the aqueous phase pH to 3.5-6.5, and purify the target product using medium-pressure preparative chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthesis route offers tangible benefits that extend far beyond the laboratory. The shift to a more robust and selective chemical process directly translates into enhanced supply chain reliability and cost efficiency. By utilizing Tenofovir Disoproxil free base as the starting material, manufacturers can leverage existing supply chains for bulk API intermediates, reducing the risk of raw material shortages associated with specialized precursors. The simplified workup procedure, which eliminates the need for complex salt-breaking steps and extensive neutralization, significantly reduces the consumption of auxiliary chemicals and solvents, leading to a leaner and more cost-effective manufacturing process.
- Cost Reduction in Manufacturing: The elimination of expensive and hazardous reagents like trifluoroacetic acid, combined with the higher selectivity of the lithium hydroxide system, drastically reduces the cost of goods sold. The improved yield means less raw material is wasted, and the simplified purification process lowers the operational expenditure associated with chromatography and solvent recovery. Furthermore, the avoidance of over-hydrolysis by-products minimizes the loss of valuable intermediate, ensuring that a higher percentage of input mass is converted into saleable, high-purity product.
- Enhanced Supply Chain Reliability: Relying on commodity chemicals such as lithium hydroxide and dichloromethane mitigates the supply risks associated with proprietary or hard-to-source reagents. The robustness of the two-phase reaction makes the process less sensitive to minor fluctuations in operating conditions, ensuring consistent batch-to-batch quality. This reliability is crucial for maintaining the continuity of supply for critical impurity standards, which are essential for the release testing of finished pharmaceutical products. A stable supply of these standards prevents bottlenecks in the QC pipeline, facilitating faster time-to-market for generic TAF formulations.
- Scalability and Environmental Compliance: The aqueous-organic two-phase system is inherently safer and easier to scale than homogeneous acidic reactions, which often require specialized corrosion-resistant equipment. The ability to separate the product into the aqueous phase simplifies isolation and reduces the volume of organic waste generated. This aligns with modern green chemistry principles, reducing the environmental footprint of the manufacturing process. For facilities operating under strict environmental regulations, this method offers a compliant pathway to produce high-value intermediates without incurring excessive waste disposal costs or requiring major infrastructure upgrades.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Tenofovir degradation impurities. These insights are derived directly from the experimental data and process optimizations detailed in the patent literature, providing a clear understanding of the method's capabilities and limitations. Understanding these nuances is vital for technical teams evaluating the feasibility of integrating this synthesis route into their existing quality control workflows.
Q: Why is Lithium Hydroxide preferred over other bases for this hydrolysis?
A: Lithium hydroxide offers superior selectivity in the two-phase system, preventing the excessive hydrolysis of active sites that occurs with stronger bases like potassium hydroxide, thereby ensuring higher purity of the target impurity standard.
Q: What are the critical active sites susceptible to hydrolysis in Tenofovir Disoproxil?
A: The molecule contains three sensitive active sites: the phenyl phosphate ester bond, the alanine ester bond, and the phosphonate linkage. Precise control is required to hydrolyze only the specific ester bond to form Compound I.
Q: How does this new method improve post-treatment compared to traditional acidic hydrolysis?
A: Unlike traditional methods using trifluoroacetic acid which form difficult-to-remove salts and cause over-hydrolysis, this alkaline two-phase method allows for simple layer separation and direct chromatographic purification, significantly simplifying the workflow.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tenofovir Disoproxil Fumarate Degradation Impurity Supplier
At NINGBO INNO PHARMCHEM, we recognize that the integrity of your pharmaceutical products depends on the quality of your reference standards. Our team of expert chemists has extensively analyzed advanced synthesis protocols, including the selective hydrolysis methods described in recent patents, to ensure we deliver the highest purity intermediates to our global partners. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that whether you need gram quantities for method development or kilogram scales for routine QC, our supply remains uninterrupted. Our state-of-the-art rigorous QC labs and commitment to stringent purity specifications guarantee that every batch of Tenofovir impurity standard meets the exacting demands of international regulatory agencies.
We invite you to collaborate with us to optimize your supply chain for Tenofovir Alafenamide quality control. By leveraging our technical expertise and manufacturing capabilities, you can secure a stable source of high-purity impurities that ensures your compliance and accelerates your product development cycles. Please contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our advanced manufacturing processes can support your strategic goals in the competitive antiviral market.