Advanced Synthesis of Tenofovir Alafenamide Degradation Impurity for Quality Control
Advanced Synthesis of Tenofovir Alafenamide Degradation Impurity for Quality Control
The pharmaceutical industry continuously demands higher standards for quality control, particularly for complex nucleotide analogs like Tenofovir Alafenamide (TAF). A recent technical breakthrough documented in patent CN113105505A introduces a highly efficient preparation method for specific degradation impurities associated with this critical antiviral agent. This innovation addresses the longstanding challenge of obtaining high-purity reference standards necessary for validating analytical methods and ensuring patient safety. By utilizing a selective biphasic hydrolysis strategy, the process achieves conversion rates exceeding 90.0% while maintaining exceptional reaction selectivity. This technical advancement is pivotal for manufacturers aiming to establish robust quality assurance protocols for TAF-based medications. The ability to reliably synthesize these degradation products allows for precise monitoring of drug stability and shelf-life.
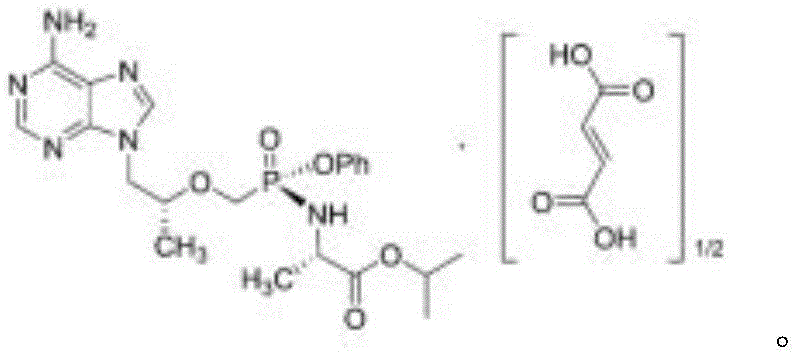
Understanding the molecular architecture of TAF is essential for grasping the complexity of its degradation pathways. The structure contains multiple hydrolytically sensitive sites, making the selective generation of specific impurities a significant synthetic challenge. The new method described leverages a deep understanding of these chemical vulnerabilities to create a controlled degradation environment. This approach not only simplifies the production of reference materials but also enhances the overall reliability of impurity profiling in commercial manufacturing settings. For global supply chains, having access to such well-characterized impurities is a cornerstone of regulatory compliance and risk management.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of TAF degradation impurities has relied on multi-step organic synthesis routes that are both cumbersome and hazardous. Prior art, such as the methods disclosed in patent CN108101942A, typically involves an initial acylation reaction followed by a separate amidation step to construct the impurity molecule from scratch. These conventional pathways suffer from several critical drawbacks, including prolonged reaction times and the necessity for handling dangerous acylating agents. Furthermore, the lack of selectivity in these traditional methods often leads to the formation of numerous byproducts, complicating the downstream purification process significantly. The cumulative effect of these inefficiencies is a substantial increase in production costs and a reduction in the overall yield of the target reference standard. Such limitations hinder the ability of quality control laboratories to rapidly obtain the necessary materials for method validation.
The Novel Approach
In stark contrast to the laborious traditional methods, the novel approach utilizes a direct hydrolysis strategy that streamlines the entire production workflow. By starting with the TAF free base itself and subjecting it to controlled alkaline conditions, the process selectively cleaves the phenol phosphate ester bond to generate the desired degradation impurity. This paradigm shift eliminates the need for constructing the molecule from smaller fragments, thereby reducing the number of unit operations and the consumption of reagents. The reaction is conducted in a biphasic system using dichloromethane and an aqueous alkaline solution, which facilitates easy separation of the product into the aqueous phase. This simplicity translates directly into operational efficiency, allowing for faster turnaround times in producing reference standards. The method represents a significant leap forward in process chemistry for pharmaceutical impurity synthesis.
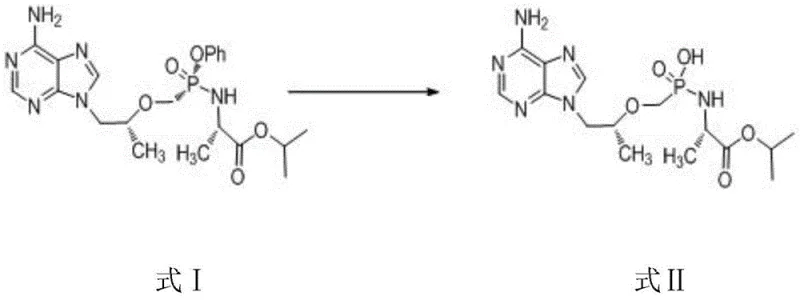
Mechanistic Insights into Selective Alkaline Hydrolysis
The success of this preparation method hinges on the precise manipulation of reaction conditions to exploit the differential reactivity of various functional groups within the TAF molecule. The core mechanism involves the nucleophilic attack of hydroxide ions, generated in situ from the alkaline substance, on the phosphorus center of the phenol phosphate ester. However, the molecule also contains an alanine ester moiety and other potential hydrolysis sites that must remain intact to ensure the identity of the target impurity. The patent data reveals that the choice of alkaline substance is the critical variable governing this selectivity. Weak bases such as potassium carbonate provide a buffered environment that promotes the hydrolysis of the more labile phenol phosphate bond while leaving the alkyl ester groups largely unaffected. This delicate balance is difficult to achieve with stronger bases, which tend to cause indiscriminate hydrolysis and degrade the molecular scaffold.
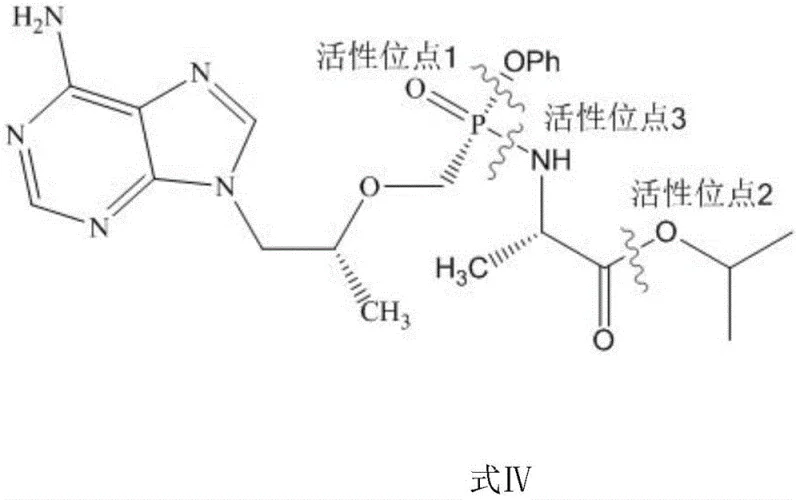
Furthermore, the control of reaction temperature and molar ratios plays a vital role in optimizing the conversion efficiency and minimizing side reactions. Experimental data indicates that maintaining the reaction temperature between 20-25°C creates an ideal kinetic profile for the selective transformation. Deviations from this range can lead to either incomplete conversion or the activation of secondary degradation pathways. The molar ratio of the TAF free base to the alkaline substance is equally important, with a ratio of 1:2.0 proving optimal for maximizing yield. This mechanistic understanding allows process chemists to fine-tune the reaction parameters to achieve consistent results across different batch sizes. Such control is essential for producing reference materials that meet the stringent purity requirements of pharmacopeial standards.
How to Synthesize Tenofovir Alafenamide Degradation Impurity Efficiently
Implementing this synthesis route in a laboratory or pilot plant setting requires adherence to specific operational parameters to ensure safety and reproducibility. The process begins with the dissolution of the TAF free base in an organic solvent, followed by the controlled addition of the aqueous alkaline phase. Strict temperature control is maintained throughout the reaction period to prevent thermal runaway or selectivity loss. Once the reaction reaches completion, indicated by the conversion of the starting material, the phases are allowed to separate. The aqueous layer, containing the target impurity as a salt, is then isolated for purification. The final step involves preparative chromatography to remove any remaining trace impurities, followed by freeze-drying to obtain the solid reference standard. Detailed standard operating procedures are essential for scaling this technology effectively.
- Dissolve Tenofovir Alafenamide free base in dichloromethane and mix with an aqueous potassium carbonate solution.
- Maintain the biphasic reaction mixture at 20-25°C with stirring to ensure selective hydrolysis of the phenol phosphate ester bond.
- Separate the aqueous layer and purify the target impurity using preparative chromatography followed by freeze-drying.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this innovative synthesis method offers compelling advantages for procurement managers and supply chain directors seeking to optimize their sourcing strategies for pharmaceutical intermediates. The elimination of complex multi-step synthesis reduces the dependency on a wide range of specialized reagents, many of which can be subject to market volatility or supply disruptions. By simplifying the raw material list to common chemicals like potassium carbonate and dichloromethane, the process enhances supply chain resilience and reduces the administrative burden of managing multiple vendors. This simplification also leads to a reduction in the overall cost of goods sold, as fewer processing steps translate to lower labor and utility consumption. Consequently, companies adopting this technology can achieve significant cost reductions in API manufacturing support activities without compromising on quality.
- Cost Reduction in Manufacturing: The streamlined nature of the direct hydrolysis process inherently lowers production costs by removing the need for expensive acylating agents and the associated waste disposal fees. Traditional methods often require stoichiometric amounts of costly reagents and generate significant chemical waste that requires treatment. In contrast, the new method utilizes catalytic or near-stoichiometric amounts of inexpensive inorganic bases, drastically cutting material costs. Additionally, the simplified workup procedure reduces the consumption of solvents and energy required for distillation and drying. These cumulative savings contribute to a more economical production model for high-purity reference standards, allowing for better margin management in competitive markets.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals rather than specialized custom synthons significantly mitigates the risk of supply chain bottlenecks. Potassium carbonate and dichloromethane are widely available from numerous global suppliers, ensuring that production can continue uninterrupted even if one vendor faces issues. This redundancy is crucial for maintaining the continuity of supply for critical quality control materials. Furthermore, the robustness of the reaction conditions means that the process is less sensitive to minor variations in raw material quality, further stabilizing the supply output. For supply chain heads, this translates to reduced lead times for high-purity pharmaceutical intermediates and greater predictability in inventory planning.
- Scalability and Environmental Compliance: The biphasic reaction system is inherently scalable, moving easily from gram-scale laboratory experiments to kilogram-scale commercial production without the need for specialized high-pressure or cryogenic equipment. This ease of scale-up facilitates rapid response to increased demand for reference materials as regulatory requirements tighten. Moreover, the process generates less hazardous waste compared to traditional methods, aligning with modern environmental, social, and governance (ESG) goals. The use of aqueous workups and the avoidance of toxic acylating agents simplify waste treatment protocols and reduce the environmental footprint of the manufacturing site. This compliance advantage is increasingly important for multinational corporations aiming to meet strict sustainability targets.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. They are derived from the specific beneficial effects and experimental data presented in the patent documentation. Understanding these details helps stakeholders evaluate the feasibility of integrating this method into their existing quality control workflows. The answers provide clarity on purity expectations, scalability, and the chemical rationale behind the process improvements. This information is vital for making informed decisions about adopting new technical standards for impurity management.
Q: Why is potassium carbonate preferred over sodium hydroxide for this hydrolysis?
A: Potassium carbonate provides superior selectivity, preventing the hydrolysis of other sensitive ester bonds in the molecule, which results in significantly higher purity compared to strong bases like sodium hydroxide.
Q: What is the expected purity of the impurity standard produced?
A: Using the disclosed chromatographic purification method, the final product achieves a purity level exceeding 99%, making it suitable for use as a certified reference substance.
Q: Is this process scalable for industrial reference material production?
A: Yes, the biphasic reaction system uses common solvents and mild conditions, allowing for straightforward scale-up from laboratory grams to multi-kilogram production batches without complex equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tenofovir Alafenamide Impurity Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-quality reference standards in the development and manufacturing of antiviral therapeutics. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements with consistency. We are committed to delivering materials that meet stringent purity specifications through our rigorous QC labs, which utilize state-of-the-art analytical instrumentation for verification. By leveraging advanced synthesis techniques like the one described in CN113105505A, we provide our partners with a competitive edge in regulatory compliance and product quality. Our dedication to technical excellence makes us a trusted partner for complex pharmaceutical intermediate needs.
We invite you to collaborate with us to optimize your supply chain for Tenofovir Alafenamide impurities and related compounds. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific production volumes and quality requirements. We encourage you to contact us to request specific COA data and route feasibility assessments for your projects. Together, we can ensure the highest standards of quality and efficiency in your pharmaceutical manufacturing operations.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →