Advanced Asymmetric Synthesis of Tenofovir Alafenamide Intermediates for Commercial Scale Production
Advanced Asymmetric Synthesis of Tenofovir Alafenamide Intermediates for Commercial Scale Production
The global demand for high-purity antiviral agents continues to surge, driven by the critical need for effective treatments against HIV and chronic hepatitis B. At the forefront of this therapeutic landscape is Tenofovir Alafenamide (TAF), a prodrug that offers superior bioavailability and safety profiles compared to its predecessor, Tenofovir Disoproxil Fumarate (TDF). A pivotal advancement in the manufacturing of this key pharmaceutical ingredient is detailed in patent CN110981911A, which discloses a highly efficient preparation method for Tenofovir Alafenamide, specifically the free base form known as GS-7340 (Formula IV). This intellectual property represents a significant leap forward in process chemistry, addressing long-standing challenges in stereochemical control and reaction kinetics that have historically plagued the industrial synthesis of nucleotide analogues.
The molecular architecture of TAF is complex, comprising a PMPA moiety, a phenoxy group, an isopropyl L-alaninate section, and a hemifumaric acid component. Traditional synthetic routes often struggle with the precise installation of the chiral phosphorus center, which is critical for the drug's biological activity. The methodology outlined in CN110981911A introduces a refined strategy that enhances the controllability of the reaction while drastically shortening the overall processing time. By decoupling the halogenation and asymmetric transformation steps, manufacturers can achieve a diastereomeric ratio (S/R) of greater than 90:10 with unprecedented efficiency. This technical breakthrough not only ensures the production of high-purity pharmaceutical intermediates but also establishes a robust foundation for reliable supply chains capable of meeting the rigorous demands of global health markets.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
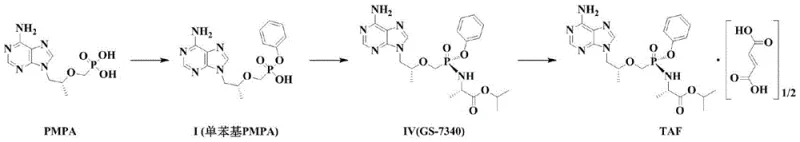
Historically, the synthesis of Formula IV from the monophenyl PMPA precursor (Formula I) has been categorized into two primary approaches, both of which present significant drawbacks for large-scale manufacturing. The first conventional method involves synthesizing a racemic mixture of the free base GS-7171, where the Rp and Sp configurations exist in an approximately 50/50 ratio. To obtain the biologically active Sp configuration (Formula IV), this mixture must undergo physical or chemical resolution, typically via chromatography or recrystallization. This approach is inherently inefficient because it theoretically caps the maximum yield at 50%, necessitating the recycling or disposal of the unwanted R-isomer. Furthermore, resolution processes are often labor-intensive, require expensive chiral columns or specific resolving agents, and introduce additional unit operations that complicate the production workflow and increase the risk of impurity accumulation.
The second category of prior art attempts to bypass resolution through direct asymmetric synthesis. For instance, existing literature describes reacting Compound I directly with thionyl chloride to generate a phosphorus oxychloride intermediate with a diastereomer ratio of S:R > 90:10. While this avoids the 50% yield loss of resolution, it suffers from severe kinetic limitations. The patent background highlights that such direct asymmetric methods require continuous reaction times ranging from 48 to 96 hours. Such prolonged exposure to reactive conditions increases energy consumption significantly and raises the probability of side reactions and product degradation. In an industrial setting, occupying reactors for up to four days for a single step creates a massive bottleneck, limiting throughput and driving up capital expenditures. These inefficiencies underscore the urgent need for a process that combines the stereo-selectivity of asymmetric synthesis with the speed required for commercial viability.
The Novel Approach
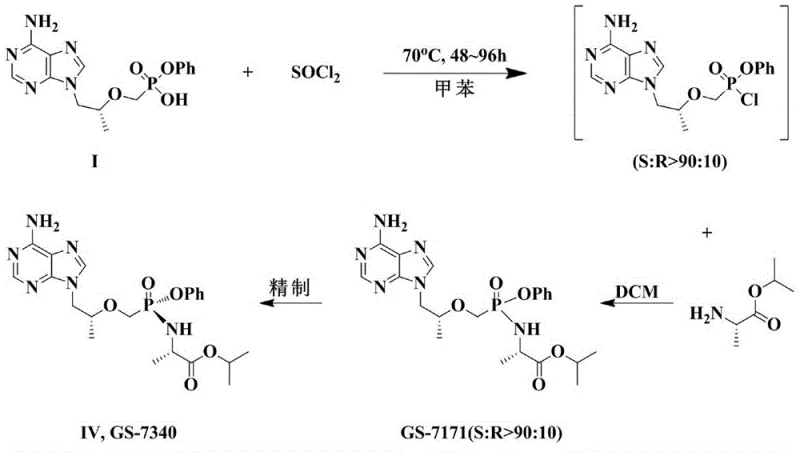
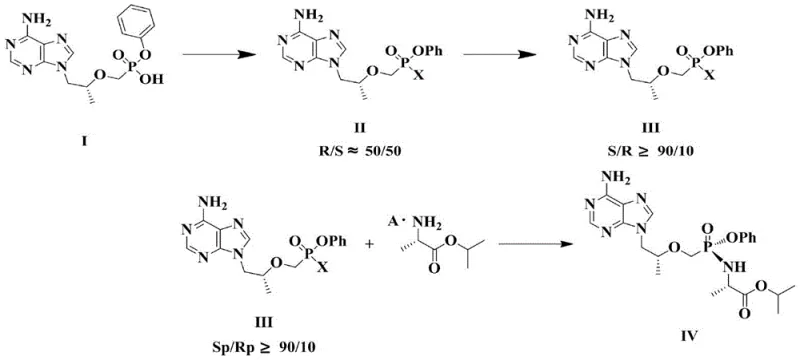
The innovation presented in CN110981911A fundamentally reengineers the synthetic pathway by splitting the transformation into distinct, optimized stages. Instead of attempting a single-pot asymmetric conversion that drags on for days, the new method first performs a rapid halogenation reaction to convert Compound I into a halogenated intermediate (Compound II). This step is straightforward and proceeds to completion quickly, ensuring that the starting material is fully activated without the extended thermal stress associated with older methods. Following this, the process employs a solvent-mediated asymmetric transformation on Compound II. By carefully selecting specific aromatic solvents and controlling the temperature below the reflux point, the system drives the equilibrium towards the desired S-isomer (Compound III) with a ratio exceeding 90:10. This strategic decoupling allows each step to be optimized independently, resulting in a total reaction time for the critical transformation phase of merely 10 to 17 hours—a reduction of over 70% compared to the 48-96 hour window of previous techniques.
Mechanistic Insights into Solvent-Mediated Asymmetric Transformation
The core of this technological advancement lies in the nuanced understanding of solvent effects on stereochemical equilibration. The patent elucidates that the asymmetric transformation of the halogenated Compound II is not merely a function of time but is critically dependent on the solvation environment. The inventors identified that aromatic hydrocarbons such as toluene, xylene, ethylbenzene, and chlorobenzene provide the ideal medium for this isomerization. Mechanistically, these solvents likely stabilize the transition state leading to the thermodynamically more stable S-diastereomer while minimizing the energy barrier for the conversion from the R-form. However, the temperature parameter is equally vital; the reaction must be conducted at elevated temperatures to accelerate kinetics but strictly maintained below the solvent's reflux temperature. Exceeding this thermal threshold disrupts the delicate equilibrium, leading to a degradation in the S/R ratio and compromising the optical purity of the final product. This precise thermal window (e.g., 85-105°C for toluene) ensures that the system reaches the desired >90% S-isomer content efficiently without triggering decomposition pathways.
Furthermore, the subsequent amidation step demonstrates a sophisticated approach to impurity control and yield maximization. The reaction between the enriched Compound III and L-isopropyl alanine is conducted at low temperatures (-10 to 10°C) in dichloromethane. This cryogenic condition is essential to suppress non-specific nucleophilic attacks that could lead to bis-amidation or hydrolysis of the sensitive phosphonate ester bonds. Following the coupling, the purification strategy employs a seeded crystallization technique using a mixed solvent system of a good solvent (like isopropanol) and a poor solvent (like isopropyl ether). By introducing seed crystals of the pure Sp-configuration, the process directs the crystallization lattice to exclusively incorporate the target enantiomer, effectively rejecting minor impurities and residual R-isomers. This "crystallization-induced dynamic resolution" effect ensures that the final high-purity pharmaceutical intermediates meet stringent HPLC specifications (often >99.5% purity) and chiral purity standards (>99.5% ee), which are non-negotiable for regulatory approval in the pharmaceutical sector.
How to Synthesize Tenofovir Alafenamide Efficiently
Implementing this novel synthesis route requires strict adherence to the optimized parameters defined in the patent to ensure reproducibility and quality. The process begins with the activation of the PMPA derivative using a halogenating agent like thionyl chloride in acetonitrile, followed by the critical solvent-mediated isomerization in toluene or xylene. The final coupling with the alanine moiety must be performed under anhydrous, low-temperature conditions to preserve the integrity of the chiral center. For process engineers and chemists looking to adopt this methodology, the following guide outlines the standardized operational sequence derived from the exemplary embodiments. It is imperative to monitor the S/R ratio via HPLC at each stage to confirm the efficacy of the asymmetric transformation before proceeding to the amidation step.
- Perform halogenation on Compound I using thionyl chloride or oxalyl chloride in acetonitrile to obtain racemic halogenated Compound II.
- Conduct solvent-mediated asymmetric transformation on Compound II in organic solvents like toluene or xylene at controlled temperatures (below reflux) to enrich the S-isomer in Compound III (S/R ≥ 90: 10).
- React Compound III with L-isopropyl alanine or its salt in dichloromethane at low temperatures (-10 to 10°C), followed by crystallization to isolate high-purity Formula IV.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis method translates into tangible strategic advantages beyond mere technical elegance. The most immediate impact is on production throughput and asset utilization. By reducing the reaction time of the key stereoselective step from nearly a week to less than a day, manufacturers can significantly increase the number of batches produced per year on existing equipment. This intensification of the process means that the same capital investment can generate a much higher volume of output, effectively lowering the fixed cost per kilogram of the intermediate. Additionally, the elimination of complex resolution steps, which often involve significant material loss and solvent consumption, streamlines the material flow and reduces the burden on waste treatment facilities. This leaner manufacturing footprint aligns perfectly with modern sustainability goals and cost-reduction initiatives.
- Cost Reduction in Manufacturing: The economic implications of this process are profound, primarily driven by the drastic reduction in cycle time and the avoidance of yield-limiting resolution steps. Conventional resolution methods inherently discard or require the recycling of half the material, which doubles the effective cost of goods sold for the raw materials. By achieving high diastereomeric excess directly through asymmetric transformation, this method maximizes atom economy. Furthermore, the shortened reaction time leads to substantial savings in utility costs, such as heating, cooling, and agitation energy. The use of common, commodity solvents like toluene and acetonitrile, rather than exotic or highly specialized reagents, ensures that raw material costs remain stable and predictable, shielding the supply chain from volatile pricing of niche chemicals.
- Enhanced Supply Chain Reliability: In the context of global pharmaceutical supply, reliability is paramount. The robustness of this synthetic route, characterized by its tolerance to standard industrial conditions and the use of readily available reagents, minimizes the risk of batch failures. Shorter cycle times also mean that inventory turnover is faster, allowing suppliers to respond more agilely to fluctuations in market demand. If a sudden surge in demand for HIV or Hepatitis B treatments occurs, a manufacturing line utilizing this 10-17 hour transformation process can ramp up output much quicker than one constrained by 96-hour reaction cycles. This agility provides a critical buffer against supply disruptions, ensuring a continuous flow of high-purity pharmaceutical intermediates to downstream formulation partners.
- Scalability and Environmental Compliance: Scaling chemical processes from the laboratory to multi-ton production often reveals hidden complexities, but this method is explicitly designed with industrial scalability in mind. The solvent-mediated transformation relies on thermodynamic equilibrium rather than sensitive catalytic systems that might deactivate on a large scale. This inherent stability simplifies the technology transfer process. Moreover, the reduction in solvent usage and the elimination of chromatographic purification steps significantly decrease the volume of hazardous waste generated. This not only lowers disposal costs but also facilitates compliance with increasingly stringent environmental regulations regarding solvent emissions and waste discharge, making it a sustainable choice for long-term commercial scale-up of complex pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and claims within patent CN110981911A, providing a clear picture of the process capabilities and limitations. Understanding these details is crucial for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: How does the new solvent-mediated asymmetric transformation improve upon previous methods?
A: Unlike previous asymmetric synthesis methods that required continuous reaction times of 48 to 96 hours, this novel approach utilizes a two-step sequence (halogenation followed by transformation) that completes the critical stereochemical enrichment in just 10 to 17 hours, significantly enhancing process controllability and throughput.
Q: What solvents are critical for achieving high diastereomeric ratios in Compound III?
A: The patent specifies that aromatic solvents such as toluene, xylene, ethylbenzene, and chlorobenzene are essential. Specifically, controlling the reaction temperature below the reflux point of these solvents (e.g., 85-105°C for toluene) is crucial to achieving an S/R ratio of greater than or equal to 90:10.
Q: Is this process suitable for large-scale industrial production of TAF intermediates?
A: Yes, the method is explicitly designed for industrial suitability. By eliminating the need for lengthy 4-day reaction cycles and avoiding complex chromatographic resolutions that waste 50% of the material, the process offers a robust pathway for commercial scale-up with improved energy efficiency and yield consistency.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tenofovir Alafenamide Supplier
As the pharmaceutical industry continues to evolve, the ability to manufacture complex antiviral intermediates with precision and efficiency is a key differentiator. NINGBO INNO PHARMCHEM stands at the forefront of this evolution, leveraging advanced synthetic methodologies like the one described in CN110981911A to deliver superior value to our global partners. Our facility is equipped with state-of-the-art reactor trains capable of handling the specific solvent and temperature requirements of this asymmetric transformation, ensuring that every batch meets the highest standards of quality. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, allowing us to seamlessly transition from clinical supply to full-scale commercial manufacturing. Our commitment to quality is underpinned by stringent purity specifications and rigorous QC labs that utilize advanced HPLC and chiral analysis to verify the S/R ratios and impurity profiles of every lot.
We invite procurement leaders and R&D directors to explore how our optimized production capabilities can enhance your supply chain resilience. By partnering with us, you gain access to a Customized Cost-Saving Analysis that quantifies the economic benefits of switching to this more efficient synthetic route. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments tailored to your project timelines. Let us collaborate to secure a reliable, cost-effective, and high-quality supply of Tenofovir Alafenamide intermediates for the global market.