Scalable Total Synthesis of D-Penicillamine: A Breakthrough in Chiral API Manufacturing
Scalable Total Synthesis of D-Penicillamine: A Breakthrough in Chiral API Manufacturing
The pharmaceutical industry continuously seeks robust, scalable, and safe manufacturing routes for critical active pharmaceutical ingredients (APIs), and the recent disclosure in patent CN111909067B represents a significant leap forward in the production of D-penicillamine. This pivotal document outlines a novel organic total synthesis method that fundamentally shifts the paradigm from traditional semi-synthetic approaches derived from penicillin to a fully controlled chemical synthesis starting from L-serine derivatives. For R&D directors and supply chain leaders, this innovation addresses long-standing concerns regarding allergen contamination, optical purity, and process scalability. By leveraging a streamlined five-step sequence involving Grignard reaction, oxidation, sulfonylation, thioesterification, and hydrolysis, the technology promises to deliver high-purity D-penicillamine with exceptional enantiomeric excess, effectively mitigating the risks associated with residual penicillin antigens that have plagued previous manufacturing methods.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the commercial production of penicillamine has relied heavily on the hydrolysis of penicillin, a semi-synthetic route that, while established, carries inherent and unacceptable risks for modern pharmaceutical standards. As illustrated in the traditional pathway below, this method involves breaking down the complex beta-lactam structure of penicillin, which often leaves trace amounts of the parent antibiotic in the final product. For patients suffering from autoimmune diseases like rheumatoid arthritis or Wilson's disease, who require long-term penicillamine therapy, even minute residues of penicillin can precipitate life-threatening hypersensitivity reactions. Furthermore, the structural complexity of penicillin makes the hydrolysis process difficult to control with precision, often resulting in variable yields and challenging purification protocols to remove structurally similar impurities. The reliance on fermentation-derived starting materials also introduces batch-to-batch variability that complicates regulatory compliance and quality assurance.
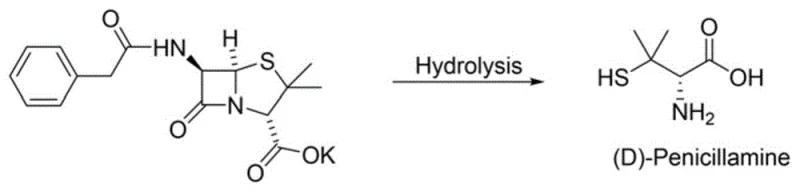
The Novel Approach
In stark contrast to the legacy hydrolysis methods, the innovative process detailed in CN111909067B utilizes a rational, step-wise construction of the penicillamine skeleton from simple, chiral building blocks. The core of this strategy is the utilization of L-serine ester derivatives, which serve as a cheap and readily available source of chirality, thereby eliminating the need for expensive chiral catalysts or resolution steps later in the synthesis. The reaction sequence is designed for operational simplicity and safety, avoiding the use of highly toxic reagents or extreme conditions that typically hinder scale-up. By constructing the molecule through a logical progression of functional group transformations—specifically installing the gem-dimethyl and thiol groups in a controlled manner—the process ensures that the stereochemical integrity of the starting material is preserved throughout. This approach not only guarantees a product free from penicillin allergens but also achieves optical purity levels (ee > 99%) that are difficult to attain via semi-synthesis, making it an ideal candidate for reliable API intermediate supplier partnerships.
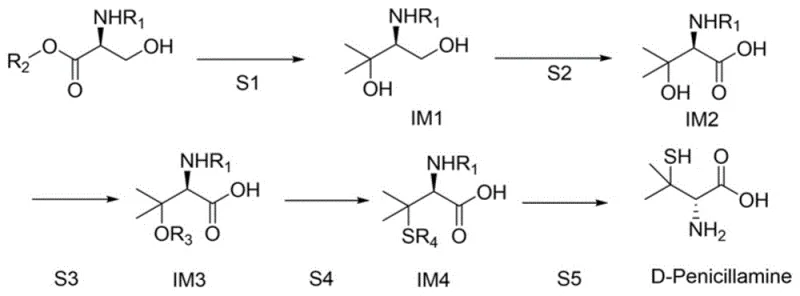
Mechanistic Insights into the Five-Step Synthetic Sequence
The chemical elegance of this total synthesis lies in its precise manipulation of functional groups to construct the sterically crowded quaternary carbon center characteristic of penicillamine. The process initiates with a Grignard reaction where a derivative of L-serine ester, such as tert-butyloxycarbonyl-L-serine methyl ester, reacts with methylmagnesium bromide at cryogenic temperatures (e.g., -30°C). This low-temperature control is critical to prevent racemization and ensure the selective formation of the tertiary alcohol intermediate without affecting the protected amine. Following this, a selective oxidation step converts the primary alcohol of the diol intermediate into a carboxylic acid using mild oxidants like bleach water in the presence of a buffer, a transformation that highlights the process's commitment to safety and environmental compatibility. The subsequent sulfonylation activates the tertiary hydroxyl group, turning a poor leaving group into an excellent one, which sets the stage for the crucial introduction of the sulfur atom.
The final stages of the synthesis demonstrate remarkable efficiency in installing the thiol functionality and revealing the final amino acid structure. The activated sulfonate intermediate undergoes a nucleophilic substitution with a sulfurization reagent, typically potassium thioacetate, to form a thioester. This indirect method of introducing sulfur is superior to using hydrogen sulfide or thiols directly, as it minimizes odor and safety hazards while providing a stable intermediate that is easy to purify. The sequence concludes with a vigorous hydrolysis step using hydrochloric acid, which simultaneously cleaves the thioester to the free thiol and removes the N-protecting group. This one-pot deprotection and hydrolysis strategy significantly reduces processing time and solvent usage. Throughout this entire mechanistic pathway, the chiral center derived from the L-serine remains intact, ensuring that the final D-penicillamine product maintains high optical purity without the formation of the toxic L-enantiomer, a critical quality attribute for regulatory approval.
How to Synthesize D-Penicillamine Efficiently
Implementing this synthesis requires careful attention to reaction parameters, particularly temperature control during the Grignard addition and pH management during the oxidation phase. The protocol described in the patent provides a robust framework for laboratory and pilot-scale execution, emphasizing the use of common solvents like THF and DCM which facilitates easy technology transfer. The detailed standardized synthetic steps below outline the specific molar ratios, temperatures, and workup procedures required to achieve the reported high yields and purity profiles. Operators should note the importance of rigorous drying of reagents for the organometallic step and the careful quenching procedures to ensure safety during scale-up operations.
- Perform a Grignard reaction on an L-serine ester derivative with methylmagnesium bromide at low temperature to form a diol intermediate.
- Oxidize the primary alcohol of the diol intermediate using an oxidant such as bleach water to obtain the corresponding carboxylic acid.
- Conduct a sulfonylation reaction on the tertiary hydroxyl group using a sulfonylation reagent like p-toluenesulfonyl chloride.
- Execute a thioesterification reaction using a sulfurization reagent such as potassium thioacetate to introduce the sulfur moiety.
- Complete the synthesis via hydrolysis reaction under acidic conditions to remove protecting groups and yield D-penicillamine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this total synthesis route offers compelling strategic advantages that extend far beyond simple chemical curiosity. The primary driver for adoption is the drastic reduction in supply chain risk associated with raw material sourcing; unlike penicillin, which is subject to fermentation bottlenecks and biological variability, L-serine derivatives are commodity chemicals produced via established industrial processes with stable pricing and abundant availability. This shift decouples penicillamine production from the volatile antibiotics market, ensuring a more predictable and continuous supply stream for downstream drug manufacturers. Furthermore, the elimination of penicillin residues removes a major regulatory hurdle, simplifying the validation process and reducing the likelihood of costly batch rejections due to allergen testing failures.
- Cost Reduction in Manufacturing: The economic viability of this process is underpinned by the use of inexpensive starting materials and the avoidance of costly chiral resolution steps. By starting with a chiral pool material like L-serine, the synthesis bypasses the need for expensive chiral catalysts or enzymatic resolutions that often plague asymmetric synthesis. Additionally, the process avoids the use of precious metal catalysts or exotic reagents, relying instead on standard organic transformations that can be performed in conventional stainless steel reactors. The high yield at each step, particularly the thioesterification and hydrolysis stages, minimizes raw material waste and maximizes the output per batch, leading to substantial cost savings in the overall cost of goods sold (COGS).
- Enhanced Supply Chain Reliability: From a logistics perspective, the stability of the intermediates and the robustness of the reaction conditions contribute to a more resilient supply chain. The intermediates, such as the sulfonate and thioester derivatives, are sufficiently stable to allow for flexible scheduling and potential storage, providing a buffer against demand fluctuations. The process does not rely on single-source biological feeds, thereby diversifying the supply base and reducing the risk of shortages caused by fermentation failures or agricultural issues affecting penicillin precursors. This reliability is crucial for maintaining uninterrupted production of finished dosage forms for chronic conditions.
- Scalability and Environmental Compliance: The environmental profile of this synthesis is significantly improved compared to traditional methods, aligning with modern green chemistry principles and stringent environmental regulations. The avoidance of heavy metal catalysts simplifies wastewater treatment and reduces the burden of hazardous waste disposal. The reaction conditions are mild, often operating near room temperature or with standard cooling, which lowers energy consumption. The simplicity of the workup procedures, involving standard extractions and crystallizations, facilitates easy scale-up from kilogram to multi-ton production without the need for specialized equipment, ensuring that the process remains economically efficient even at very large commercial volumes.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis route. These insights are derived directly from the experimental data and comparative analysis presented in the patent documentation, aiming to clarify the operational benefits and quality improvements offered by this technology. Understanding these nuances is essential for technical teams evaluating the feasibility of adopting this new manufacturing standard.
Q: Why is total synthesis preferred over penicillin hydrolysis for D-penicillamine?
A: Traditional semi-synthesis via penicillin hydrolysis carries a significant risk of penicillin residue, which can trigger severe allergic reactions in sensitive patients. Total synthesis eliminates this biological contamination risk entirely while offering superior control over optical purity.
Q: What are the key advantages of using L-serine derivatives as starting materials?
A: L-serine derivatives, such as tert-butyloxycarbonyl-L-serine methyl ester, are commercially available, cost-effective, and possess high inherent optical purity. This allows the synthesis to proceed without complex chiral induction steps, significantly simplifying the process and reducing purification costs.
Q: How does this new process impact scalability and environmental safety?
A: The process utilizes mild reaction conditions and avoids toxic heavy metal catalysts or hazardous reagents often found in older methods. The simplicity of the five-step sequence, combined with high yields at each stage, makes it highly amenable to large-scale commercial production with reduced environmental burden.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable D-Penicillamine Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this total synthesis technology for the global pharmaceutical market. As a premier CDMO partner, we possess the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. Our state-of-the-art facilities are equipped to handle the specific requirements of this synthesis, including cryogenic reactions and sensitive sulfur chemistry, while our rigorous QC labs enforce stringent purity specifications to guarantee that every batch meets the highest international standards for optical purity and impurity profiles.
We invite forward-thinking pharmaceutical companies to collaborate with us to leverage this advanced manufacturing route for their D-penicillamine supply needs. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how this new process can optimize your budget. We encourage you to contact us today to request specific COA data and route feasibility assessments, allowing you to make informed decisions that enhance both the quality and reliability of your supply chain.