Advanced Synthesis of Natural Glycosyl-1-Phosphoryl Dipeptides for Commercial Scale-Up
Introduction to Next-Generation Phosphoglycopeptide Synthesis
The landscape of pharmaceutical intermediate manufacturing is constantly evolving, driven by the need for higher purity and more efficient synthetic routes for complex bioactive molecules. Patent CN103435688A introduces a groundbreaking methodology for the preparation of natural glycosyl-1-phosphoryl dipeptides and phosphoramides, specifically targeting compounds like Phosphoramidone and Tarotide. These molecules are critical research tools and potential therapeutic agents known for their potent inhibition of thermolysin and endothelin-converting enzymes, which are pivotal in hypertension and renal disease pathways. The disclosed technology addresses long-standing challenges in the field, such as the scarcity of rare sugar sources like 6-deoxytalose and the inefficiency of traditional phosphorylation reagents. By leveraging a novel phosphorous tri-imidazole intermediate and a refined oxidative coupling system, this invention promises to transform the supply chain for these high-value biochemical reagents. For R&D directors and procurement specialists, understanding this shift is essential for securing reliable sources of high-purity pharmaceutical intermediates that meet stringent quality specifications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the chemical synthesis of phosphoramidone and its analogues has been plagued by significant technical hurdles that hinder commercial viability. Early reported methods, such as the direct condensation of α-L-rhamnose-1-phosphate monoester with dipeptides, suffered from abysmal yields of approximately 6%, rendering them impractical for any scale beyond milligram-level research. Alternative strategies employing phenyl dichlorophosphate or iodine-mediated oxidation of phosphite monoesters offered marginal improvements but still struggled with yields hovering around 44% to 64%. A major bottleneck in these legacy processes is the propensity for forming anomeric mixtures and the requirement for specialized, often unstable, phosphitylating reagents that complicate inventory management. Furthermore, the final deprotection steps frequently involved harsh saponification conditions that degraded the sensitive phosphoamide bond, leading to substantial material loss and difficult purification profiles. These inefficiencies translate directly into exorbitant costs and unreliable supply chains for downstream drug development projects relying on these key intermediates.
The Novel Approach
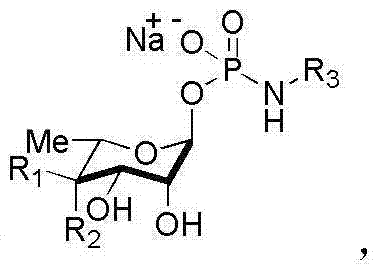
In stark contrast, the methodology outlined in the patent data presents a robust, three-step sequence designed to maximize throughput and minimize waste. The core innovation lies in the initial generation of a glycosyl-1-hydrogen phosphite monoester using phosphorous tri-imidazole, which dramatically improves the initial phosphorylation efficiency to over 90%. This is followed by a sophisticated oxidative coupling step utilizing a carbon tetrachloride and triethylamine system, which generates a highly reactive glycosyl-1-phosphoryl chloride intermediate in situ. This specific intermediate facilitates a cleaner coupling reaction with various amino compounds, pushing precursor yields to exceed 80%. The final stage employs a gentle one-pot hydrogenation and deacetylation protocol that avoids the decomposition issues associated with strong bases, achieving final product yields of greater than 90%. This holistic optimization ensures that the production of complex structures, as illustrated in the general formula below, becomes economically feasible and technically reproducible on a multi-kilogram scale.
Mechanistic Insights into Phosphitylation and Oxidative Coupling
The success of this synthetic route hinges on the precise control of phosphorus chemistry during the initial activation of the sugar moiety. The reaction begins with the formation of phosphorous tri-imidazole from phosphorus trichloride and imidazole, a reagent that offers superior nucleophilic displacement capabilities compared to standard chlorinating agents. When reacted with triacetyl-protected rhamnose or 6-deoxytalose at temperatures ranging from -20°C to 40°C, this reagent selectively forms the glycosyl-1-hydrogen phosphite monoester with high stereocontrol. The subsequent silylation using N,O-bis(trimethylsilyl)acetamide (BSA) protects the acidic proton, preventing unwanted side reactions during the critical oxidation phase. This careful orchestration of reagents ensures that the phosphorus atom remains in the correct oxidation state prior to the final coupling, thereby eliminating the formation of pyrophosphate byproducts that often contaminate batches produced via older methods.
Furthermore, the oxidative coupling mechanism represents a significant departure from traditional iodine-based systems, which are prone to over-oxidation and coloration issues. By utilizing a carbon tetrachloride and triethylamine system, the process generates a glycosyl-1-phosphoryl chloride intermediate that is exceptionally electrophilic yet stable enough to be handled in situ. This intermediate reacts efficiently with leucine-tryptophan dipeptide benzyl esters or other amines at mild temperatures between 0°C and 30°C. The mechanistic advantage here is the avoidance of radical pathways that can degrade the peptide backbone, ensuring the integrity of the chiral centers within the dipeptide portion. This level of chemical fidelity is paramount for producing high-purity pharmaceutical intermediates where even trace impurities can alter biological activity or toxicity profiles in preclinical studies.
How to Synthesize Glycosyl-1-Phosphoryl Dipeptides Efficiently
Implementing this synthesis in a GMP environment requires strict adherence to the reaction parameters defined in the patent to ensure consistent quality and yield. The process is divided into three distinct operational phases: the formation of the phosphite monoester, the oxidative coupling to form the protected precursor, and the final global deprotection. Each step utilizes common industrial solvents such as dichloromethane, tetrahydrofuran, and methanol, facilitating easy solvent recovery and recycling. The detailed standardized synthesis steps below outline the specific molar ratios, temperature controls, and workup procedures necessary to replicate the high yields reported in the intellectual property documentation. Following these guidelines allows manufacturers to bypass the trial-and-error phase typically associated with scaling up complex glycosylation reactions.
- React phosphorus trichloride with imidazole to form phosphorous tri-imidazole, then couple with protected rhamnose or talose to obtain glycosyl-1-hydrogen phosphite monoester.
- Perform silylation with BSA followed by oxidation with carbon tetrachloride/triethylamine to generate a glycosyl-1-phosphoryl chloride intermediate for coupling with dipeptides.
- Execute a one-pot hydrogenation using palladium on carbon and deacetylation with sodium methoxide to remove protecting groups and purify the final target compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthesis route offers tangible benefits that extend far beyond simple yield improvements. The elimination of expensive and hazardous reagents like molecular iodine significantly reduces the raw material cost base and simplifies waste disposal protocols. Traditional methods often required specialized purification steps to remove iodine residues and colored impurities, which added both time and expense to the manufacturing cycle. By switching to the carbon tetrachloride/triethylamine oxidation system, the process generates cleaner crude products that require less intensive chromatographic purification, thereby reducing the consumption of silica gel and eluents. This streamlining of the downstream processing directly contributes to cost reduction in pharmaceutical intermediate manufacturing, allowing for more competitive pricing models without sacrificing margin.
- Cost Reduction in Manufacturing: The primary driver for cost savings in this new method is the drastic improvement in overall yield across all three synthetic steps. While legacy routes might lose up to 80% of material during deprotection due to decomposition, this new protocol preserves over 90% of the intermediate mass. Additionally, the use of phosphorous tri-imidazole eliminates the need for custom-synthesized phosphitylating agents that often carry high price tags and long lead times. The ability to use commodity chemicals like phosphorus trichloride and imidazole as starting materials stabilizes the supply chain against market volatility. Consequently, the total cost of goods sold (COGS) is significantly lowered, making the final active ingredients more affordable for end-users in the biotech sector.
- Enhanced Supply Chain Reliability: Supply continuity is often threatened by the reliance on rare natural sources for starting materials, such as the microbial extraction of 6-deoxytalose. This synthetic method circumvents that bottleneck by enabling the total chemical synthesis of the tarotide scaffold from readily available protected sugars. The robustness of the reaction conditions, which tolerate a range of temperatures and solvent grades, means that production is less susceptible to minor fluctuations in utility availability or reagent quality. This resilience ensures that delivery schedules for high-purity pharmaceutical intermediates can be met consistently, reducing the risk of project delays for clients developing enzyme inhibitors or antihypertensive therapies.
- Scalability and Environmental Compliance: Scaling chemical processes from gram to kilogram levels often exposes hidden inefficiencies, but this method has been validated for preparation at the 5-gram level with clear pathways for expansion. The one-pot deprotection strategy minimizes the number of unit operations, reducing the equipment footprint and energy consumption required for heating and cooling cycles. Furthermore, by avoiding heavy metal catalysts in the oxidation step and utilizing palladium on carbon which can be recovered and recycled, the environmental impact is substantially mitigated. This alignment with green chemistry principles facilitates easier regulatory approval and supports the sustainability goals of modern chemical enterprises seeking to reduce their carbon footprint.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and benefits of this patented synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on how this method outperforms historical precedents. Understanding these nuances is critical for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios. The answers highlight the specific chemical advantages that translate into commercial value for partners seeking a reliable pharmaceutical intermediate supplier.
Q: How does this new method improve yield compared to conventional iodine oxidation?
A: Conventional methods utilizing iodine oxidation often suffer from yields around 44% due to side reactions and anomer formation. The novel approach described in patent CN103435688A utilizes a carbon tetrachloride/triethylamine system to generate a specific glycosyl-1-phosphoryl chloride intermediate, boosting the precursor yield to over 80% while minimizing impurities.
Q: What are the advantages of the deprotection strategy for large-scale production?
A: Traditional saponification methods can lead to product decomposition under strong alkaline conditions, limiting yields to 20-30%. This patent introduces a mild one-pot hydrogenation and deacetylation protocol using palladium catalysts and controlled sodium methoxide addition, achieving yields exceeding 90% and ensuring stability for commercial batch sizes.
Q: Is this synthesis route suitable for generating diverse phosphoramidone analogues?
A: Yes, the modular nature of the coupling step allows for the introduction of various amino compounds (H2N-R3), including dipeptides like Leu-Trp and cyclic amines like morpholine. This flexibility supports the rapid generation of libraries for structure-activity relationship studies in drug discovery programs targeting metalloproteases.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Phosphoramidone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the successful development of life-saving medications. Our team of expert chemists has extensively analyzed the technological breakthroughs presented in patent CN103435688A and possesses the capability to execute this advanced synthesis with precision. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met whether you are in the early discovery phase or full-scale manufacturing. Our facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of glycosyl-1-phosphoryl dipeptide meets the highest industry standards for potency and impurity profiles.
We invite you to collaborate with us to leverage this cutting-edge technology for your next project. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis that details how switching to this novel route can optimize your budget. We encourage you to contact us today to discuss your specific requirements,索取 specific COA data, and review our comprehensive route feasibility assessments. Let us help you secure a stable, cost-effective supply of these complex molecules, empowering your research and development efforts to reach new heights of success.