Advanced Organosilicon Synthesis: Streamlining Pharmaceutical Intermediate Production via Direct C-H Activation
The landscape of organic synthesis is undergoing a paradigm shift towards more sustainable and atom-economical processes, particularly in the realm of pharmaceutical intermediate manufacturing. Patent CN112778352B, published in September 2022, introduces a groundbreaking methodology for the synthesis of novel organosilicon compounds via palladium-catalyzed direct C-H activation. This technology represents a significant departure from classical substitution reactions, offering a streamlined pathway to access structurally diverse aryl silanes directly from commercially available acetophenone derivatives. For R&D directors and process chemists, this patent outlines a robust protocol that leverages a specific oxime-based directing group to achieve high ortho-selectivity, bypassing the need for harsh pre-functionalization steps that have historically plagued silicon-carbon bond formation. The implications for supply chain efficiency and cost reduction in fine chemical production are substantial, as this one-pot transformation consolidates multiple synthetic operations into a single, efficient catalytic cycle.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the synthesis of organosilicon compounds has relied heavily on the reaction of organometallic intermediates, such as Grignard or organolithium reagents, with silicon electrophiles, or alternatively, transition metal-catalyzed cross-coupling of aryl halides with hydrosilanes. These conventional pathways suffer from inherent inefficiencies, primarily due to the requirement for pre-functionalization of the aromatic substrate. Converting a stable C-H bond into a reactive C-X bond (where X is a halogen or pseudohalogen) necessitates additional synthetic steps, generates stoichiometric amounts of salt waste, and often requires cryogenic conditions or highly reactive reagents that pose safety hazards on a large scale. Furthermore, achieving specific regioselectivity, particularly at the ortho-position relative to a carbonyl group, often demands complex protecting group strategies or suffers from poor selectivity profiles, leading to difficult purification challenges and reduced overall yields in commercial manufacturing settings.
The Novel Approach
In stark contrast, the methodology disclosed in CN112778352B utilizes a direct C-H activation strategy that fundamentally alters the retrosynthetic logic for accessing these valuable intermediates. By employing a palladium catalyst in conjunction with a carefully designed oxime directing group, the process enables the direct installation of a trimethylsilyl group onto the ortho-position of acetophenone derivatives. This approach eliminates the need for pre-halogenated starting materials, thereby significantly improving the atom economy of the transformation. The reaction proceeds under relatively mild thermal conditions using hexamethyldisilane as the silicon source, which is both commercially accessible and safer to handle than many chlorosilane alternatives. This novel route not only simplifies the synthetic sequence but also enhances the environmental profile of the manufacturing process by reducing the generation of halogenated waste streams, aligning perfectly with modern green chemistry principles demanded by regulatory bodies and corporate sustainability goals.
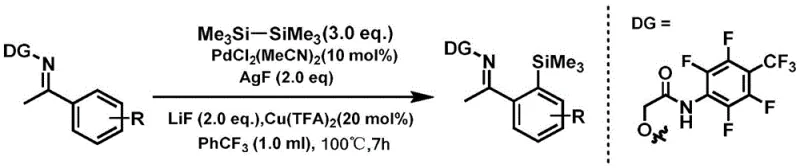
Mechanistic Insights into Pd-Catalyzed Ortho-C-H Silylation
The core of this technological advancement lies in the intricate interplay between the palladium catalyst and the bidentate oxime directing group attached to the acetophenone substrate. The mechanism likely initiates with the coordination of the palladium(II) species to the nitrogen and oxygen atoms of the oxime moiety, forming a stable cyclopalladated intermediate. This coordination brings the metal center into close proximity with the ortho-C-H bond, facilitating its cleavage through a concerted metalation-deprotonation (CMD) pathway or a sigma-bond metathesis mechanism. The presence of additives such as silver fluoride (AgF) and copper trifluoroacetate (Cu(TFA)2) plays a critical dual role: they act as oxidants to regenerate the active palladium species and potentially assist in the transmetallation step with the hexamethyldisilane reagent. The use of lithium fluoride as a base further promotes the deprotonation event, ensuring high turnover frequencies. This precise orchestration of catalytic components allows for the selective formation of the C-Si bond while leaving other sensitive functional groups on the aromatic ring intact, a feature that is paramount for the synthesis of complex pharmaceutical scaffolds where orthogonal reactivity is essential.
From an impurity control perspective, the high regioselectivity imparted by the directing group is a major advantage for process chemistry. In non-directed C-H functionalization, mixtures of ortho-, meta-, and para-isomers are common, requiring energy-intensive chromatographic separations that are impractical at multi-kilogram scales. Here, the steric and electronic constraints imposed by the palladacycle ensure that silylation occurs almost exclusively at the desired ortho-position. Furthermore, the choice of trifluorotoluene as the solvent provides a high-boiling, non-coordinating medium that stabilizes the cationic palladium intermediates without interfering with the catalytic cycle. The subsequent workup involves simple filtration through silica gel to remove insoluble silver and copper salts, followed by standard column chromatography, indicating a straightforward isolation protocol that minimizes product loss and maximizes recovery rates, which is crucial for maintaining cost-effectiveness in commercial production.
How to Synthesize Ortho-Silylated Acetophenone Derivatives Efficiently
The practical implementation of this synthesis involves a straightforward one-pot procedure that is amenable to standard laboratory and pilot plant equipment. The process begins with the dissolution of the acetophenone oxime substrate, hexamethyldisilane, and the catalytic system comprising PdCl2(MeCN)2, AgF, Cu(TFA)2, and LiF in trifluorotoluene. The reaction mixture is then heated in a sealed pressure vessel to ensure the retention of volatile components and to maintain the necessary pressure for efficient mass transfer at elevated temperatures. Following the reaction period, the mixture is cooled, and the heterogeneous catalyst residues are removed via filtration, yielding a crude solution that can be directly subjected to purification. This operational simplicity reduces the technical barrier for adoption and allows for rapid screening of substrate scope.
- Prepare the reaction mixture by dissolving the acetophenone derivative containing the oxime directing group, hexamethyldisilane (3.0 equivalents), PdCl2(MeCN)2 catalyst, AgF oxidant, Cu(TFA)2 co-catalyst, and LiF base in trifluorotoluene solvent.
- Seal the mixture in a thick-walled pressure tube and heat to 100°C under oil bath stirring for approximately 7 hours to facilitate the C-H activation and silylation.
- Upon completion, cool the reaction, filter through silica gel to remove insoluble salts, wash with ethyl acetate, and purify the crude product via column chromatography to obtain the target organosilicon compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this C-H activation technology offers tangible benefits regarding raw material sourcing and process scalability. By utilizing widely available acetophenone derivatives as starting materials, the supply chain becomes less dependent on specialized, pre-functionalized building blocks that often have limited suppliers and long lead times. The elimination of halogenated precursors not only reduces the cost of goods sold (COGS) associated with purchasing expensive aryl halides but also mitigates the logistical and disposal costs associated with handling and treating halogenated waste. This shift towards more abundant feedstocks enhances supply chain resilience, ensuring continuity of supply even during market fluctuations for specialty reagents. Additionally, the robustness of the catalytic system suggests that the process can be scaled from gram to kilogram quantities with minimal re-optimization, facilitating a smoother transition from R&D to commercial manufacturing.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the high atom economy and the reduction in synthetic steps. By avoiding the pre-functionalization of the aromatic ring, manufacturers save on the reagents and solvents typically required for halogenation and subsequent protection/deprotection sequences. Although specific percentage savings depend on the specific substrate, the qualitative reduction in material consumption is significant. The use of hexamethyldisilane, a bulk chemical, instead of specialized silylating agents further contributes to lower input costs. Moreover, the simplified purification workflow reduces the consumption of silica gel and eluents, which are often hidden cost drivers in fine chemical production.
- Enhanced Supply Chain Reliability: Reliance on commodity chemicals like acetophenones and hexamethyldisilane ensures a stable supply base. Unlike complex chiral ligands or exotic catalysts that may face supply bottlenecks, the reagents used in this patent are produced by multiple global vendors. This diversification of the supply base reduces the risk of production stoppages due to single-source failures. The mild reaction conditions (100°C) also imply that the process does not require specialized high-pressure or cryogenic reactors, allowing it to be manufactured in a wider range of existing facilities, thereby increasing overall production capacity and flexibility.
- Scalability and Environmental Compliance: The environmental footprint of this synthesis is markedly lower than traditional methods. The absence of stoichiometric halogenated byproducts simplifies wastewater treatment and reduces the burden on effluent processing units. As regulatory pressures on pharmaceutical and chemical manufacturers intensify regarding waste discharge and carbon emissions, adopting greener C-H activation technologies provides a strategic advantage. The process generates fewer solid wastes (primarily metal salts which can be recovered or treated) and avoids the use of volatile organic solvents associated with Grignard-type reactions, aligning with stringent environmental, health, and safety (EHS) standards required for GMP manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this patented technology. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing clarity on the reaction's versatility and limitations. Understanding these nuances is critical for process development teams evaluating this route for potential integration into their existing manufacturing pipelines.
Q: What is the primary advantage of this C-H silylation method over traditional routes?
A: The primary advantage is the elimination of pre-functionalization steps. Traditional methods often require converting C-H bonds to C-X bonds (like halides) before silylation, which generates stoichiometric waste. This patent describes a direct C-H activation strategy that improves atom economy and reduces synthetic steps.
Q: Can the trimethylsilyl group introduced by this method be further functionalized?
A: Yes, the patent demonstrates that the trimethylsilyl group serves as a versatile handle. It can be efficiently converted into other functional groups, such as aryl iodides via iodination, which subsequently allows for cross-coupling reactions like Suzuki coupling to build complex biaryl structures.
Q: What specific directing group is required for this ortho-selective silylation?
A: The method utilizes an oxime-based directing group installed on the acetophenone substrate. This specific group coordinates with the palladium catalyst to direct the activation specifically to the ortho-position of the aromatic ring, ensuring high regioselectivity.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Organosilicon Compound Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced C-H activation technologies in accelerating drug discovery and process development. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative academic protocols like those described in CN112778352B can be successfully translated into robust industrial processes. We are committed to delivering high-purity organosilicon intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Whether you require custom synthesis of complex silylated building blocks or optimization of existing routes for cost and efficiency, our CDMO capabilities are designed to support your most challenging projects.
We invite you to collaborate with us to explore how this novel silylation methodology can enhance your product portfolio. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific project needs. We are ready to provide specific COA data and comprehensive route feasibility assessments to help you make informed decisions about integrating these advanced organosilicon compounds into your supply chain.