Scalable Synthesis of A-76202 Analogs: Advanced Suzuki Coupling for High-Purity Pharmaceutical Intermediates
Introduction to Advanced Chromone Glycoside Synthesis
The pharmaceutical industry continuously seeks robust synthetic pathways for bioactive flavonoid derivatives, particularly those exhibiting potent alpha-glucosidase inhibitory activity. Patent CN101279993B introduces a sophisticated methodology for producing 3-aryl-8-hydroxychromone-7-O-alpha-D-arabinofuranoside compounds, which serve as powerful analogs of the natural product A-76202. These compounds are critical intermediates in the development of therapeutics for diabetes, obesity, and viral infections. The disclosed technology overcomes historical challenges in flavonoid glycosylation by employing a late-stage functionalization strategy. 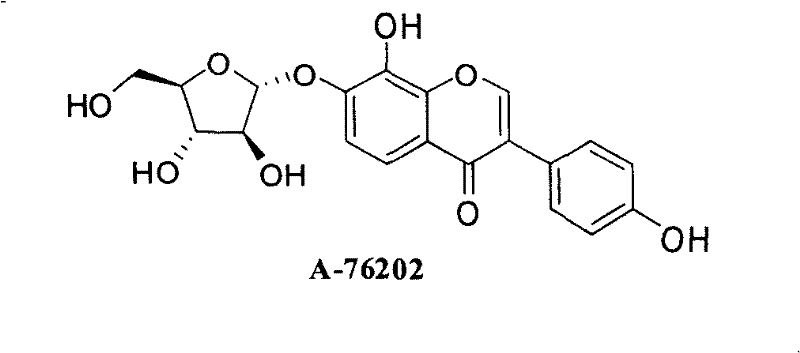 By decoupling the construction of the chromone core from the installation of the diverse B-ring aryl group, this process offers unprecedented flexibility for medicinal chemists aiming to optimize structure-activity relationships (SAR).
By decoupling the construction of the chromone core from the installation of the diverse B-ring aryl group, this process offers unprecedented flexibility for medicinal chemists aiming to optimize structure-activity relationships (SAR).
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthesis of complex flavonoid glycosides often relies on early-stage glycosylation or direct arylation methods that suffer from poor regioselectivity and harsh reaction conditions. Conventional routes frequently struggle to differentiate between the multiple hydroxyl groups present on the chromone scaffold, leading to complex mixtures of regioisomers that are difficult and costly to separate. Furthermore, introducing the aryl B-ring prior to glycosylation can deactivate the substrate towards subsequent sugar attachment due to steric hindrance or electronic effects. These inefficiencies result in low overall yields, excessive solvent waste, and significant bottlenecks in scaling up production for clinical trials.
The Novel Approach
The methodology described in the patent revolutionizes this landscape by utilizing a modular assembly line. Instead of attempting to build the entire molecule in one pot, the process isolates the synthesis of the glycosylated chromone core first. 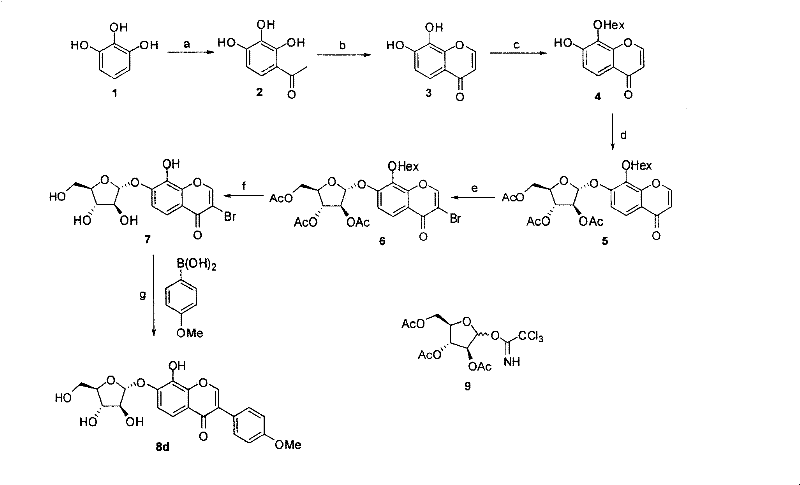 This intermediate is then brominated at the 3-position, creating a versatile handle for palladium-catalyzed cross-coupling. This strategic inversion of the synthetic sequence ensures that the sensitive glycosidic bond is established under optimized conditions before the final diversification step. The result is a cleaner reaction profile, simplified purification protocols, and a platform technology capable of generating a library of analogs with minimal process changes.
This intermediate is then brominated at the 3-position, creating a versatile handle for palladium-catalyzed cross-coupling. This strategic inversion of the synthetic sequence ensures that the sensitive glycosidic bond is established under optimized conditions before the final diversification step. The result is a cleaner reaction profile, simplified purification protocols, and a platform technology capable of generating a library of analogs with minimal process changes.
Mechanistic Insights into Pd-Catalyzed Suzuki-Miyaura Coupling
The cornerstone of this synthetic innovation is the final Suzuki-Miyaura coupling reaction, which constructs the carbon-carbon bond between the chromone core and the variable aryl ring. The patent specifies the use of Palladium(II) acetate (Pd(OAc)2) in conjunction with the SPhos ligand. SPhos (2-dicyclohexylphosphino-2',6'-dimethoxybiphenyl) is a bulky, electron-rich biaryl phosphine ligand known for accelerating oxidative addition and facilitating reductive elimination. In the context of these electron-deficient chromone bromides, the SPhos ligand stabilizes the active Pd(0) species, preventing catalyst aggregation and ensuring high turnover numbers even at moderate temperatures of 50°C.
Impurity control is meticulously managed through the preceding bromination step. By using PhI(OAc)2 and TMSBr to generate active molecular bromine in situ, the process achieves high selectivity for the 3-position of the chromone ring. This precision is vital because bromination at other positions would lead to inactive byproducts that could co-elute with the desired API intermediate. The subsequent deprotection of acyl groups using potassium carbonate in a mixed aqueous solvent system further demonstrates the robustness of the glycosidic linkage, which remains intact under basic hydrolysis conditions, ensuring the final product retains its stereochemical integrity.
How to Synthesize 3-aryl-8-hydroxychromone-7-O-alpha-D-arabinofuranoside Efficiently
The synthesis begins with the cyclization of phloroglucinol derivatives to form the 7,8-dihydroxychromone core, followed by selective protection strategies to expose the 7-hydroxyl group for glycosylation. Once the sugar moiety is attached and the 3-position is brominated, the stage is set for the final diversification. The detailed standardized synthetic steps, including precise stoichiometry, temperature profiles, and workup procedures for each transformation, are outlined below to ensure reproducibility and safety in a GMP environment.
- Synthesize the 7,8-dihydroxychromone core via cyclization of acetylated phloroglucinol derivatives.
- Perform selective deprotection of the 7-hydroxyl group using hexanoyl protection and transesterification.
- Execute glycosylation at the 7-position followed by bromination at the 3-position to prepare the coupling partner.
- Conduct the final Suzuki-Miyaura coupling with aryl boronic acids using Pd(OAc)2 and SPhos ligand.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition from extraction-based sourcing to this fully synthetic route represents a paradigm shift in risk management and cost structure. The reliance on natural isolation for A-76202 analogs historically introduced volatility regarding raw material availability, seasonal variations, and batch-to-batch consistency. By adopting this chemical synthesis pathway, manufacturers can secure a stable, year-round supply of high-purity intermediates independent of biological sources. This stability is crucial for long-term drug development projects where supply continuity is a regulatory requirement.
- Cost Reduction in Manufacturing: The synthetic route eliminates the need for expensive chromatographic separations typically associated with purifying natural product extracts. The use of commodity chemicals such as phloroglucinol, acetic acid, and widely available aryl boronic acids drives down the raw material cost base significantly. Furthermore, the high yields reported in the patent examples, such as the 75% yield in the final coupling step, minimize material loss and reduce the cost of goods sold (COGS). The ability to recycle solvents like acetone and dichloromethane further enhances the economic viability of the process.
- Enhanced Supply Chain Reliability: The modularity of the Suzuki coupling step means that supply chain disruptions for one specific aryl boronic acid do not halt the entire production line; the facility can simply switch to producing a different analog within the same series using the same core intermediate. This flexibility allows for agile response to market demands. Additionally, the reagents used, such as Pd(OAc)2 and SPhos, are commercially available from multiple global suppliers, mitigating the risk of single-source dependency for critical catalysts.
- Scalability and Environmental Compliance: The process utilizes aqueous acetone mixtures for the final coupling, reducing the reliance on purely organic solvent systems and aligning with green chemistry principles. The reaction conditions (50°C) are mild and energy-efficient, requiring less heating input compared to traditional high-temperature reflux methods. This lower energy footprint, combined with the potential for telescoping steps (combining multiple reactions without isolation), simplifies the engineering requirements for scale-up from pilot plant to commercial tonnage production.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the scalability, purity, and regulatory compliance of this synthetic method. Understanding these nuances is essential for technical teams evaluating this technology for integration into their existing manufacturing portfolios. The answers are derived directly from the experimental data and claims presented in the patent documentation.
Q: What is the key advantage of using SPhos ligand in this synthesis?
A: The SPhos ligand significantly enhances the efficiency of the Suzuki coupling reaction, allowing for milder conditions (50°C) and high yields (up to 75%) even with sterically hindered substrates.
Q: How is regioselectivity achieved during the glycosylation step?
A: Regioselectivity is controlled by initially protecting both phenolic hydroxyls with a hexanoyl group, followed by selective transesterification to expose only the 7-hydroxyl group for glycosidic bond formation.
Q: Can this route be adapted for different aryl substituents?
A: Yes, the modular nature of the Suzuki coupling allows for the introduction of various aryl groups (e.g., methoxy, fluoro, dimethylamino) by simply changing the boronic acid reactant.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-aryl-8-hydroxychromone-7-O-alpha-D-arabinofuranoside Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of having a dependable partner for complex pharmaceutical intermediates. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from lab-scale discovery to market launch is seamless. We adhere to stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of A-76202 analogs meets the highest quality standards required for clinical and commercial applications.
We invite you to contact our technical procurement team to discuss how we can tailor this synthetic route to your specific volume and budget requirements. Request a Customized Cost-Saving Analysis today, and let us provide you with specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can accelerate your drug development timeline while optimizing your overall project economics.