Scalable Synthesis of A-76202 Analogues via Advanced Suzuki-Miyaura Coupling for Commercial API Production
Scalable Synthesis of A-76202 Analogues via Advanced Suzuki-Miyaura Coupling for Commercial API Production
The pharmaceutical landscape for metabolic disorders and oncology continues to demand highly potent enzyme inhibitors with optimized pharmacokinetic profiles. Patent CN101279993A introduces a sophisticated synthetic methodology for producing 3-aryl-8-hydroxychromone-7-O-α-D-arabinofuranoside compounds, which serve as powerful analogues of the natural product A-76202. These compounds exhibit strong inhibitory activity against α-glucosidase I and II, positioning them as critical candidates for treating diabetes, obesity, and even HIV infections. The structural integrity of these molecules relies heavily on the specific stereochemistry of the arabinofuranoside moiety, as depicted in the core structure below, which is essential for maintaining high biological potency compared to other glycosidic variants.
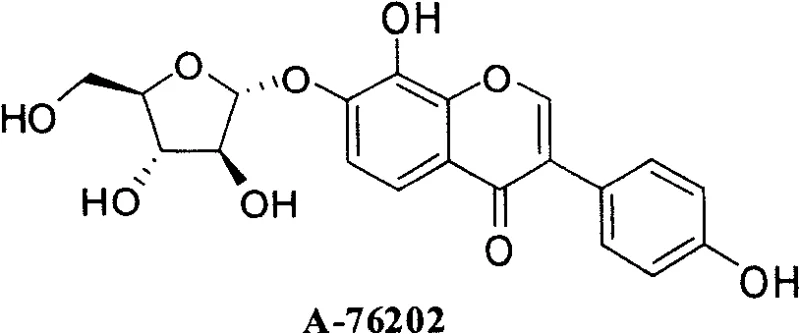
For procurement managers and supply chain directors, understanding the synthetic accessibility of such complex glycosides is paramount. The patent outlines a robust, convergent strategy that separates the construction of the chromone core from the installation of the diverse aryl B-ring. This modularity is a game-changer for cost reduction in pharmaceutical intermediate manufacturing, as it allows producers to stock a single advanced intermediate and react it with various inexpensive boronic acids to generate a library of potential drug candidates. This approach not only accelerates lead optimization but also simplifies inventory management for a reliable pharmaceutical intermediate supplier aiming to support early-stage clinical trials.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
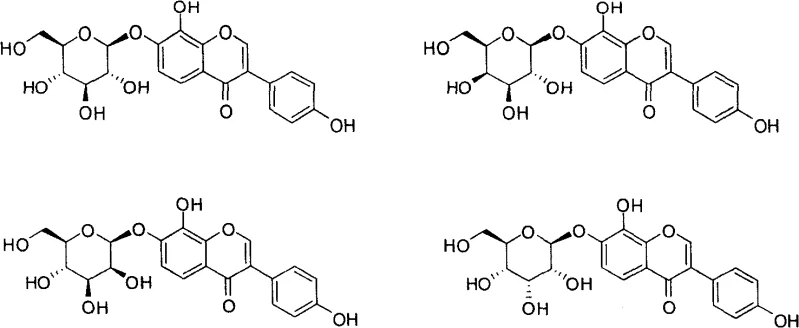
Historically, the synthesis of flavonoid glycosides like A-76202 has been plagued by regioselectivity issues and low overall yields. Early work by Shiozaki and others demonstrated that while total synthesis was possible, modifying the B-ring often required rebuilding the entire flavonoid skeleton from scratch, a tedious and resource-intensive process. Furthermore, previous attempts to modify the sugar moiety, such as synthesizing six-carbon pyranoside analogues, resulted in compounds with significantly diminished biological activity, highlighting the unforgiving nature of structure-activity relationships in this class of molecules. Traditional methods often relied on harsh conditions for glycosylation or difficult-to-control halogenation steps that generated significant impurity profiles, complicating downstream purification and increasing the cost of goods sold.
The Novel Approach
The methodology described in CN101279993A overcomes these hurdles by employing a late-stage functionalization strategy centered on the Suzuki-Miyaura cross-coupling reaction. By first constructing a 3-bromo-chromone glycoside intermediate, the inventors created a versatile platform where the aryl group can be introduced in the final steps under mild conditions. This decouples the complex glycosylation chemistry from the aromatic substitution, ensuring that the sensitive sugar moiety is not exposed to harsh conditions for longer than necessary. The use of specific protecting group strategies, such as the hexanoyl group for selective 7-OH exposure, ensures high regioselectivity during glycosylation, thereby maximizing the yield of the desired α-anomer and minimizing the formation of inactive β-isomers or regioisomers.
Mechanistic Insights into Pd-Catalyzed Suzuki-Miyaura Coupling
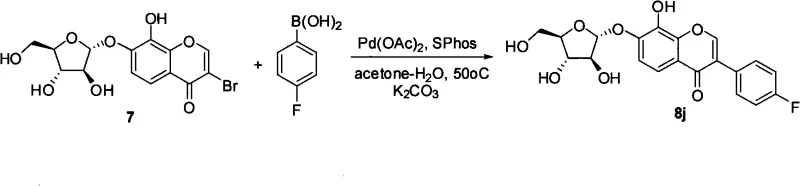
The heart of this synthetic innovation lies in the efficient construction of the C-C bond between the chromone core and the aryl ring. The patent specifies the use of Palladium(II) acetate [Pd(OAc)2] in conjunction with the bulky, electron-rich phosphine ligand SPhos. This catalyst system is particularly effective for coupling sterically hindered substrates, which is often the case when attaching substituted phenyl rings to the 3-position of the chromone. The catalytic cycle begins with the oxidative addition of the aryl bromide intermediate to the Pd(0) species, followed by transmetallation with the arylboronic acid activated by the base (K2CO3 or Na2CO3). The use of an aqueous acetone solvent system facilitates the solubility of both the organic substrate and the inorganic base, promoting efficient turnover.
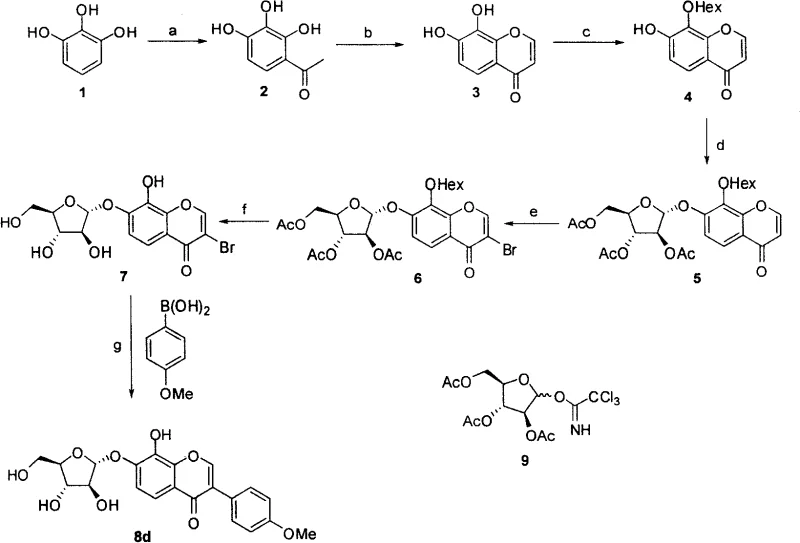
Furthermore, the preceding bromination step utilizes a sophisticated reagent system comprising PhI(OAc)2 and TMSBr to generate active molecular bromine in situ. This approach avoids the safety hazards and handling difficulties associated with elemental bromine, making the process more amenable to commercial scale-up of complex pharmaceutical intermediates. The electrophilic bromination occurs selectively at the C-3 position of the chromone ring due to the electron density distribution influenced by the carbonyl group and the glycosidic oxygen. Following the coupling reaction, the removal of acyl protecting groups under mild basic conditions (K2CO3 in MeOH-THF-H2O) reveals the free hydroxyl groups necessary for biological activity without degrading the sensitive glycosidic bond, ensuring a high-purity final product suitable for biological testing.
How to Synthesize 3-aryl-8-hydroxychromone Derivatives Efficiently
The synthesis protocol detailed in the patent provides a clear roadmap for producing these high-value intermediates. The process begins with the acylation of pyrogallol and subsequent cyclization to form the chromone core, followed by a carefully orchestrated sequence of protection, glycosylation, and bromination. The final Suzuki coupling step is performed at a moderate temperature of 50°C, which balances reaction rate with thermal stability of the glycoside. For R&D teams looking to replicate or scale this chemistry, the detailed experimental section offers precise molar ratios and solvent systems that have been optimized for reproducibility. The standardized synthetic steps see below guide ensures consistent quality across batches.
- Synthesize the 7,8-dihydroxychromone core via acylation of pyrogallol followed by cyclization with triethyl orthoformate.
- Perform selective 7-O-glycosylation using a D-arabinose donor and BF3·OEt2 promoter after protecting the 8-OH group.
- Execute bromination at the C-3 position using PhI(OAc)2/TMSBr, followed by deprotection and Suzuki coupling with arylboronic acids.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the synthetic route outlined in this patent offers substantial strategic advantages for sourcing and manufacturing. The modularity of the Suzuki coupling step means that a manufacturer can produce a single bulk quantity of the bromo-intermediate and then diversify it into multiple analogues based on demand. This flexibility drastically reduces the risk associated with holding inventory for specific drug candidates that may fail in early development. Additionally, the reagents used, such as arylboronic acids, are widely available commodity chemicals, ensuring a stable and resilient supply chain that is not dependent on exotic or hard-to-source starting materials.
- Cost Reduction in Manufacturing: The elimination of the need to rebuild the chromone core for every new analogue significantly lowers the raw material and labor costs associated with library synthesis. By utilizing a convergent synthesis where the complex sugar-containing core is prepared once and coupled with various aryl groups, the overall process mass intensity is improved. The use of relatively inexpensive catalysts and ligands, combined with high-yielding steps like the initial acylation (reported up to 96%), contributes to a more economical production process that enhances margins for generic API manufacturers.
- Enhanced Supply Chain Reliability: The reliance on robust, well-understood chemical transformations such as Suzuki coupling and standard ester hydrolysis ensures that the process can be transferred between different manufacturing sites with minimal tech transfer friction. The avoidance of cryogenic conditions or highly unstable intermediates means that the process is less susceptible to disruptions caused by equipment failures or utility fluctuations. This reliability is crucial for maintaining continuous supply to pharmaceutical clients who require consistent quality for their clinical trials and eventual commercial launches.
- Scalability and Environmental Compliance: The process utilizes solvent systems like acetone-water and dichloromethane, which are standard in the industry and can be efficiently recovered and recycled. The mild reaction temperatures (mostly 0°C to 50°C) reduce energy consumption compared to processes requiring prolonged reflux at high temperatures. Furthermore, the in situ generation of brominating agents minimizes the storage and transport hazards of toxic reagents, aligning with modern environmental, health, and safety (EHS) standards required by top-tier pharmaceutical companies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these A-76202 analogues. The answers are derived directly from the experimental data and technical specifications provided in the patent documentation, ensuring accuracy for decision-makers evaluating this technology for their pipeline.
Q: What is the key advantage of the Suzuki coupling method in this patent?
A: The Suzuki coupling allows for modular construction of the B-ring, enabling rapid diversification of the molecule with various aryl groups using commercially available boronic acids, significantly streamlining the SAR study process.
Q: Why is the arabinofuranoside moiety critical for biological activity?
A: Previous studies indicated that replacing the arabinofuranoside with hexopyranosides resulted in significantly reduced α-glucosidase inhibitory activity, making the specific furanoside configuration essential for potency.
Q: Is the bromination step safe for large-scale manufacturing?
A: Yes, the process utilizes PhI(OAc)2 and TMSBr to generate active molecular bromine in situ, avoiding the direct handling and storage hazards associated with elemental bromine gas or liquid.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-aryl-8-hydroxychromone Supplier
At NINGBO INNO PHARMCHEM, we recognize the immense potential of α-glucosidase inhibitors in the treatment of metabolic diseases and are fully equipped to support the development of these promising candidates. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to clinical supply is seamless. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of 3-aryl-8-hydroxychromone intermediates meets the highest international standards for safety and efficacy.
We invite you to contact our technical procurement team to discuss how we can tailor this synthetic route to your specific needs. Whether you require a Customized Cost-Saving Analysis for a specific analogue or need specific COA data and route feasibility assessments for your regulatory filings, we are ready to provide the data-driven insights necessary to move your project forward. Partner with us to leverage our expertise in complex glycoside synthesis and secure a reliable supply chain for your next-generation therapeutics.