Advanced Enantioselective Synthesis of Diazepinoindolones for Commercial Scale-up
Advanced Enantioselective Synthesis of Diazepinoindolones for Commercial Scale-up
The pharmaceutical industry continuously seeks robust synthetic routes for complex heterocyclic scaffolds, particularly those serving as key intermediates for potent therapeutic agents. Patent CN1362960A introduces a groundbreaking methodology for the preparation of enantiomerically pure substituted [1,4]-diazepino[6,7,1-hi]-indole-4-ones, which are critical precursors for Phosphodiesterase 4 (PDE4) inhibitors. These inhibitors hold immense potential in treating inflammatory diseases, allergies, bronchial dilation issues, and asthma. The core innovation lies in a novel cyclization strategy that bypasses the traditional and costly resolution of racemic mixtures, directly accessing the desired S-configuration enantiomers through a catalytic process. This technical advancement represents a significant leap forward in process chemistry, offering a streamlined pathway that aligns perfectly with the rigorous demands of modern Good Manufacturing Practice (GMP) standards for high-purity active pharmaceutical ingredients.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art, specifically referenced in international application WO98/49169, relied heavily on synthesizing these diazepinoindolone compounds as racemic mixtures. This approach necessitated downstream separation techniques to isolate the biologically active enantiomer, typically involving chiral phase chromatography or diastereomeric salt formation with enantiomerically pure amines. These conventional resolution methods are inherently inefficient, often resulting in a maximum theoretical yield of only 50% for the desired isomer, with the remainder being waste or requiring complex recycling streams. Furthermore, chiral chromatography is notoriously expensive and difficult to scale, consuming vast quantities of solvents and specialized stationary phases, which drastically inflates the cost of goods sold (COGS) and creates significant bottlenecks in supply chain continuity for commercial manufacturing.
The Novel Approach
In stark contrast, the method disclosed in CN1362960A utilizes an optically active 3-aminobenzodiazepine starting material that already possesses the desired stereochemistry. By employing a specific cyclization reaction catalyzed by a weak Lewis acid, the process constructs the final tetracyclic core while rigorously maintaining the integrity of the chiral center. This direct asymmetric synthesis eliminates the need for any resolution step, theoretically doubling the yield relative to racemic processes and removing the capital expenditure associated with chiral separation equipment. The ability to proceed from a chiral amine directly to the final cyclized product with high fidelity transforms the economic model of producing these intermediates, shifting the focus from separation science to efficient catalytic transformation.
Mechanistic Insights into Scandium Triflate-Catalyzed Cyclization
The heart of this invention is the careful selection of the catalyst to drive the intramolecular cyclization of the imidate intermediate (Formula II) to the final indolone (Formula I). The patent explicitly identifies weak Lewis acids, such as scandium triflate (Sc(OTf)3), as the optimal catalysts for this transformation. The mechanism involves the activation of the imidate carbonyl or imine functionality by the Lewis acid, rendering it susceptible to nucleophilic attack by the adjacent aromatic ring or amine nitrogen, thereby closing the ring. Crucially, the mild Lewis acidity prevents the protonation or coordination that would lead to the formation of a planar carbocation intermediate, which is the primary pathway for racemization or epimerization at the sensitive C-3 stereocenter.
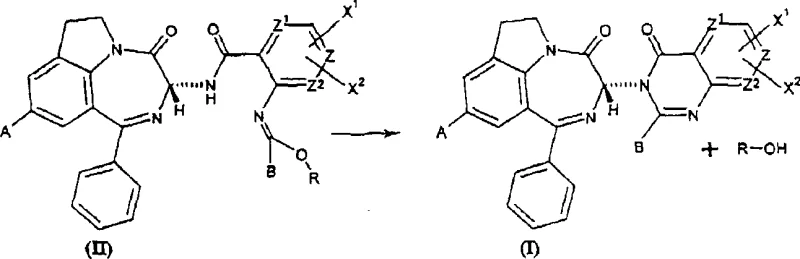
Experimental data within the patent underscores the critical nature of catalyst strength. For instance, Example 6 demonstrates that while scandium triflate yields 92% of the desired cyclic product with high optical purity, stronger Lewis acids like aluminum bromide (AlBr3) cause complete epimerization, destroying the chiral value of the molecule. Similarly, aluminum chloride and boron trifluoride show significantly lower conversion rates or higher byproduct formation. This mechanistic understanding allows process chemists to fine-tune reaction conditions, typically maintaining temperatures between 0°C and 40°C, to ensure that the kinetic pathway favors cyclization over thermodynamic equilibration that would erode enantiomeric excess. The use of inert, aprotic solvents like dichloromethane further supports this delicate balance by minimizing side reactions.
How to Synthesize Chiral Diazepinoindolones Efficiently
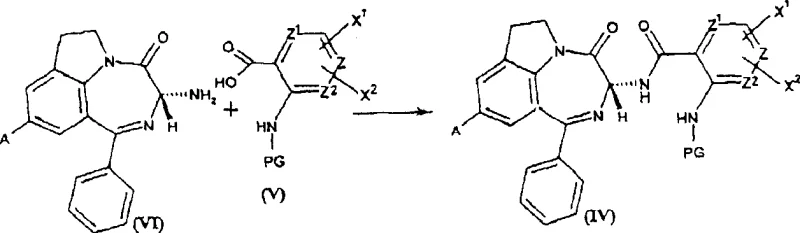
The synthesis of these high-value intermediates follows a logical sequence designed to preserve chirality at every stage. The process begins with the coupling of an optically active aminobenzodiazepine with a protected anthranilic acid derivative, followed by deprotection to generate a free amine. This amine is then converted into an imidate using an orthoester, setting the stage for the final ring closure. The detailed operational parameters, including specific molar ratios, solvent choices, and workup procedures, are critical for reproducing the high yields and purity reported in the patent examples. Understanding the nuances of each step, from the choice of coupling agents like DCC/HOBT to the vacuum distillation of methanol during imidate formation, is essential for successful technology transfer.
- Condense optically active aminobenzodiazepine with protected anthranilic acid using coupling agents like DCC/HOBT to form the amide intermediate.
- Deprotect the amide intermediate using trifluoroacetic acid to reveal the free primary amine group necessary for cyclization.
- React the free amine with an orthoester, such as trimethyl orthoacetate, under vacuum to form the imidate precursor.
- Perform the final cyclization using a catalytic amount of a weak Lewis acid like scandium triflate at controlled temperatures below 40°C to maintain stereochemistry.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the shift from a resolution-based process to a direct enantioselective synthesis offers profound strategic benefits beyond simple chemistry. The elimination of chiral chromatography or salt formation steps removes a major variable cost driver, significantly reducing the consumption of expensive chiral reagents and solvents. This simplification of the manufacturing workflow translates directly into a more robust and predictable supply chain, as the process relies on standard, commercially available achiral reagents and catalysts rather than specialized separation media that may have long lead times or single-source dependencies.
- Cost Reduction in Manufacturing: The removal of the resolution step effectively doubles the theoretical yield of the active isomer from the starting chiral material, providing a massive leverage point for cost optimization. Additionally, the patent notes that the Lewis acid catalyst can potentially be recycled at the end of the reaction, further lowering the raw material cost per kilogram. By avoiding the low-yield bottlenecks of traditional resolution, manufacturers can achieve substantial cost savings in pharmaceutical intermediate manufacturing without compromising on quality.
- Enhanced Supply Chain Reliability: The reliance on mild reaction conditions, typically near room temperature or slightly elevated, reduces the energy footprint and safety risks associated with cryogenic or high-pressure processes. This makes the process easier to implement across multiple manufacturing sites, diversifying supply risk. The starting materials, such as substituted anthranilic acids and benzodiazepines, are well-established commodity chemicals, ensuring that raw material availability remains stable even during market fluctuations.
- Scalability and Environmental Compliance: The use of weak Lewis acids and standard organic solvents simplifies waste stream management compared to processes involving heavy metals or harsh mineral acids. The ability to run the reaction at concentrations that do not require extreme dilution improves reactor throughput, facilitating the commercial scale-up of complex pharmaceutical intermediates. This environmental and operational efficiency aligns with modern green chemistry initiatives, reducing the overall ecological impact of the production lifecycle.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. They are derived from the specific experimental findings and comparative data presented in the patent documentation, providing clarity on the practical aspects of adopting this technology for industrial production.
Q: Why are weak Lewis acids preferred over strong acids in this synthesis?
A: Strong Lewis acids like aluminum bromide can cause complete epimerization of the stereocenter, leading to racemic mixtures. Weak Lewis acids like scandium triflate facilitate cyclization while preserving the high optical purity of the starting material.
Q: What is the typical optical purity achieved with this method?
A: The process consistently yields products with high enantiomeric excess (ee), often exceeding 94% to 99%, as confirmed by chiral HPLC analysis without the need for subsequent resolution steps.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the reaction conditions are mild, typically operating between 0°C and 40°C, and the catalyst can potentially be recycled, making it highly scalable and safer for industrial production compared to cryogenic or high-pressure alternatives.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Diazepinoindolone Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the synthetic route described in CN1362960A for the production of next-generation PDE4 inhibitors. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from laboratory bench to full-scale manufacturing is seamless. Our facilities are equipped with stringent purity specifications and rigorous QC labs capable of monitoring enantiomeric excess with high precision, guaranteeing that every batch meets the exacting standards required for clinical and commercial applications.
We invite you to collaborate with our technical team to evaluate how this optimized synthesis can enhance your project's economics and timeline. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the specific financial benefits applicable to your volume requirements. We encourage you to contact our technical procurement team today to obtain specific COA data and route feasibility assessments tailored to your unique development needs.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →