Scalable Manufacturing of High-Purity Empagliflozin Intermediates via Novel Stereoselective Reduction
Scalable Manufacturing of High-Purity Empagliflozin Intermediates via Novel Stereoselective Reduction
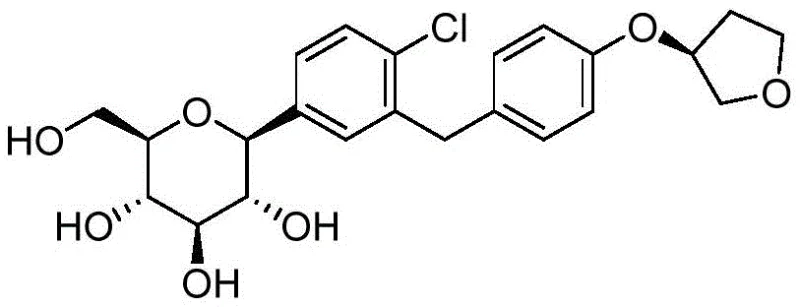
The pharmaceutical landscape for Type 2 diabetes treatment has been revolutionized by SGLT2 inhibitors, with Empagliflozin standing out as a cornerstone therapy due to its proven cardiovascular benefits. However, the industrial production of this complex molecule faces significant challenges regarding stereochemical control and impurity management. Patent CN108794548B introduces a groundbreaking methodology for preparing Englitazone (Empagliflozin) and its key intermediates, specifically addressing the critical bottleneck of isomer impurity control. This technical disclosure outlines a robust synthetic route that leverages specialized Grignard reagents and mild reduction conditions to achieve exceptional purity profiles. By integrating these advanced chemical strategies, manufacturers can overcome the limitations of traditional synthesis, ensuring a reliable supply of high-quality active pharmaceutical ingredients (APIs) for the global market.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for Empagliflozin often struggle with the precise control of anomeric stereochemistry during the coupling of the sugar moiety with the aglycone. Conventional methods frequently rely on harsh acidic conditions or non-selective catalysts that promote the formation of thermodynamic byproducts, specifically the beta-anomer and other structural isomers. These impurities are notoriously difficult to separate due to their similar physicochemical properties, often requiring multiple, yield-depleting recrystallization steps or complex chromatographic separations. Furthermore, older protocols may involve unstable intermediates or reagents that pose safety risks on a large scale, such as uncontrolled exotherms during Grignard additions. The accumulation of these isomeric impurities not only complicates the purification process but also jeopardizes the final drug substance's compliance with stringent pharmacopeial standards, leading to increased production costs and extended lead times.
The Novel Approach
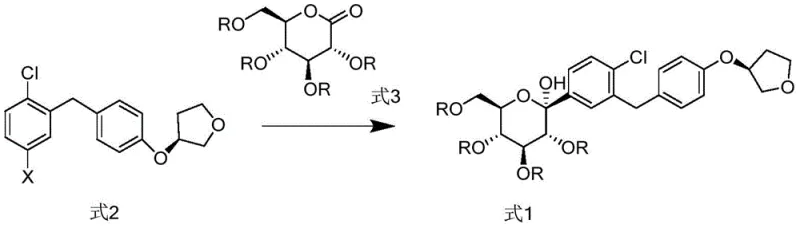
The methodology described in CN108794548B presents a paradigm shift by introducing a highly controlled two-step sequence involving a specialized Grignard reagent and a subsequent stereoselective reduction. The process initiates with the reaction of a specific aryl halide precursor with sec-butylmagnesium chloride lithium chloride, a complex that offers enhanced stability and reactivity compared to simple Grignard reagents. This is immediately followed by coupling with a protected gluconolactone at cryogenic temperatures (-50°C), which kinetically favors the formation of the desired alpha-C-glycosidic bond. Crucially, the subsequent reduction step employs a Lewis acid-silane system (Boron Trifluoride and Triethylsilane) under mild conditions, which effectively locks the stereochemistry and minimizes epimerization. This novel pathway drastically simplifies the impurity profile, allowing for the direct isolation of intermediates with isomer levels well below critical thresholds without the need for excessive purification.
Mechanistic Insights into Stereoselective Reduction and Impurity Control
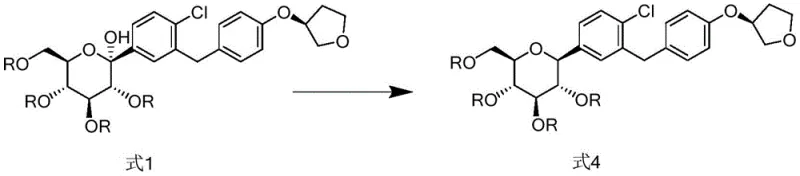
The core innovation of this process lies in the mechanistic precision of the reduction step, where the conversion of the hemiketal intermediate (Formula 1) to the C-glycoside (Formula 4) is meticulously managed. The use of Boron Trifluoride (BF3) as a Lewis acid activates the anomeric center, facilitating the hydride attack from Triethylsilane. This specific reagent combination creates a transient oxocarbenium ion-like transition state that is sterically directed to favor the alpha-configuration. By maintaining the reaction temperature strictly between -10°C and 0°C, the kinetic control is maximized, preventing the equilibration to the more stable but undesired beta-isomer. This mechanistic finesse ensures that the resulting compound of Formula 4 contains no more than 3.5% of Isomer-1 and a negligible amount (less than 0.1%) of Isomer-2, a level of purity that is exceptionally difficult to achieve with standard reducing agents like sodium borohydride or catalytic hydrogenation.
Furthermore, the process demonstrates remarkable robustness in managing the deprotection phase, which is often a source of degradation in carbohydrate chemistry. The patent specifies the use of sodium methoxide in a mixed solvent system of tetrahydrofuran and methanol. This basic condition is sufficiently mild to cleave the acetyl protecting groups without inducing beta-elimination or anomerization side reactions. The avoidance of aqueous conditions during this step is particularly advantageous, as water can promote hydrolysis of the sensitive C-glycosidic bond or facilitate mutarotation. The result is a final crude product with a relative area purity exceeding 98%, which can be further upgraded to over 99.5% through a simple slurry process. This high degree of selectivity throughout the synthetic tree underscores the process's suitability for GMP manufacturing environments where consistency and quality are paramount.
How to Synthesize Empagliflozin Intermediates Efficiently
The synthesis of these high-value intermediates requires strict adherence to the optimized parameters outlined in the patent to ensure reproducibility and safety. The process begins with the preparation of the Grignard reagent under an inert nitrogen atmosphere, followed by the critical low-temperature coupling with the lactone. Operators must ensure precise thermal management, particularly during the exothermic addition phases, to maintain the integrity of the stereochemical outcome. The subsequent reduction and deprotection steps are designed to be telescoped or performed with minimal workup, streamlining the overall workflow. For a detailed breakdown of the specific reagent quantities, solvent choices, and operational timelines required to execute this synthesis successfully, please refer to the standardized guide below.
- Prepare tetra-O-acetyl-beta-D-gluconolactone by reacting gluconolactone with acetic anhydride and trifluoroacetic acid at 80°C.
- Perform a Grignard reaction using sec-butylmagnesium chloride lithium chloride with a chloro-iodo-benzene derivative at -10 to -5°C, followed by addition to the lactone at -50°C.
- Execute a stereoselective reduction of the resulting hemiketal intermediate using triethylsilane and boron trifluoride at -10 to 0°C to minimize isomer impurities.
- Complete the synthesis via deprotection using sodium methoxide in a THF-methanol solvent system to yield the final Empagliflozin API.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this patented synthesis route offers substantial strategic advantages for procurement managers and supply chain directors seeking to optimize their API sourcing strategies. The primary value driver is the significant reduction in processing complexity, which directly translates to lower manufacturing costs and improved throughput. By eliminating the need for extensive chromatographic purification or multiple recrystallizations to remove stubborn isomers, the process reduces solvent consumption, waste generation, and equipment occupancy time. This efficiency gain allows for a more agile production schedule, enabling suppliers to respond faster to market demand fluctuations without compromising on quality standards. Additionally, the use of commercially available and relatively inexpensive reagents like triethylsilane and sodium methoxide ensures that raw material costs remain stable and predictable.
- Cost Reduction in Manufacturing: The streamlined nature of this synthetic route eliminates several costly unit operations typically associated with C-glycoside synthesis. By achieving high stereochemical selectivity directly in the reactor, the process removes the financial burden of expensive chiral separation technologies or yield-loss-heavy purification steps. The ability to perform the deprotection without aqueous workup further reduces the energy costs associated with drying and solvent recovery. Consequently, the overall cost of goods sold (COGS) for the intermediate is drastically lowered, providing a competitive pricing advantage for downstream API manufacturers.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions, particularly the tolerance for standard industrial solvents like toluene and acetonitrile, ensures that the supply chain is not vulnerable to shortages of exotic or highly regulated reagents. The process is designed to be scalable from laboratory to commercial tonnage without significant re-engineering, guaranteeing a consistent supply of intermediates. The high purity of the crude product minimizes the risk of batch failures during final API synthesis, thereby securing the continuity of the finished drug product supply and reducing the risk of stockouts for pharmaceutical partners.
- Scalability and Environmental Compliance: This methodology aligns perfectly with modern green chemistry principles by minimizing waste and maximizing atom economy. The reduction in solvent usage and the elimination of heavy metal catalysts (often used in alternative coupling methods) simplify the environmental compliance landscape. The process generates less hazardous waste, lowering disposal costs and reducing the environmental footprint of the manufacturing facility. This sustainability profile is increasingly critical for multinational corporations aiming to meet their corporate social responsibility (CSR) goals while maintaining efficient large-scale production capabilities.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthesis route. Understanding these details is crucial for R&D teams evaluating the feasibility of technology transfer and for quality assurance professionals establishing control strategies. The answers provided are derived directly from the experimental data and embodiments disclosed in the patent documentation, ensuring accuracy and relevance to real-world manufacturing scenarios.
Q: How does this process control isomer impurities in Empagliflozin synthesis?
A: The process utilizes a specific reduction protocol involving boron trifluoride and triethylsilane at low temperatures (-10 to 0°C), which effectively suppresses the formation of Isomer-1 (keeping it under 3.5%) and Isomer-2 (under 0.1%), ensuring superior stereochemical purity compared to conventional methods.
Q: What are the critical reaction conditions for the Grignard step?
A: The Grignard reaction requires strict temperature control between -10°C and -5°C during the addition of sec-butylmagnesium chloride lithium chloride, followed by a subsequent reaction with the lactone intermediate at cryogenic temperatures of -45°C to -50°C to ensure optimal yield and selectivity.
Q: Why is sodium methoxide preferred for the deprotection step?
A: Sodium methoxide is selected for the deprotection reaction because it operates under mild conditions (20-30°C) and avoids the use of water, which significantly reduces the generation of hydrolysis-related impurities and simplifies the downstream purification process.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Empagliflozin Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-purity intermediates in the production of life-saving medications like Empagliflozin. Our state-of-the-art facilities are equipped to handle complex organic syntheses, including the stereoselective reductions and Grignard couplings described in CN108794548B. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive a consistent and reliable supply of materials. Our rigorous QC labs enforce stringent purity specifications, utilizing advanced analytical techniques to monitor isomer levels and ensure every batch meets the highest industry standards before it leaves our site.
We invite global pharmaceutical partners to collaborate with us to leverage this advanced technology for their supply chains. By partnering with our technical team, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality needs. We encourage you to contact our technical procurement team today to request specific COA data, route feasibility assessments, and samples to evaluate how our optimized Empagliflozin intermediate process can enhance your operational efficiency and product quality.