Advanced Synthesis of L-735,524 Intermediates via Amide Enolate Alkylation for Commercial Scale-up
Introduction to Next-Generation HIV Protease Inhibitor Synthesis
The pharmaceutical industry continuously seeks more efficient pathways for producing complex antiretroviral agents, particularly HIV protease inhibitors like L-735,524. Patent CN1130380A discloses a groundbreaking methodology that fundamentally alters the synthetic landscape for these critical therapeutics. This technology introduces a novel approach for preparing key epoxide intermediates through the reaction of amide enolates with activated non-racemic glycidyl derivatives. Unlike traditional methods that rely on lengthy sequences and hazardous reagents, this invention enables the direct construction of the carbon backbone with high stereocontrol. For R&D directors and process chemists, this represents a significant leap forward in synthetic efficiency, offering a route that bypasses the need for toxic osmium tetroxide and reduces the overall step count dramatically. The ability to generate these complex intermediates in fewer steps directly translates to improved process mass intensity and reduced environmental impact, aligning with modern green chemistry principles while maintaining the rigorous purity standards required for API manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methods for synthesizing L-735,524 and related compounds were notoriously cumbersome, often requiring up to 12 distinct reaction steps to reach the final target. These legacy processes typically depended on the use of alkylated hydroxy-protected dihydro-5(S)-hydroxymethyl-3(2H)-furanones, which are expensive and difficult to source in bulk quantities. Furthermore, a critical bottleneck in these older routes was the necessity of using osmium tetroxide (OsO4) for dihydroxylation steps. OsO4 is not only highly toxic and expensive but also poses severe safety and waste disposal challenges on a commercial scale. The multi-step nature of these conventional syntheses inevitably led to cumulative yield losses, increased solvent consumption, and prolonged production timelines, making cost reduction in API manufacturing a significant challenge for supply chain managers.
The Novel Approach
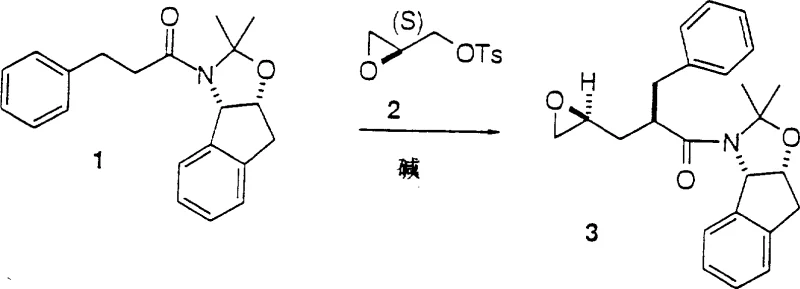
The innovative strategy outlined in the patent data circumvents these historical bottlenecks by employing a direct coupling reaction between an amide enolate and an activated glycidyl derivative. This method allows for the introduction of the three-carbon glycidyl unit in a single, highly selective step, effectively replacing multiple transformations found in previous routes. By utilizing activated species such as glycidyl p-toluenesulfonate, the process achieves high yields without the need for toxic heavy metal oxidants. This streamlined approach not only simplifies the operational complexity but also enhances the overall economic viability of the synthesis. For procurement teams, this shift means relying on more readily available starting materials and avoiding the supply chain volatility associated with specialized, hazardous reagents, thereby ensuring greater continuity and reliability in the production of high-purity pharmaceutical intermediates.
Mechanistic Insights into Amide Enolate Alkylation
The core of this technological advancement lies in the precise generation and reaction of the amide enolate. The process begins with the metallation of the amide carbonyl alpha-position using strong, non-nucleophilic bases such as lithium bis(trimethylsilyl)amide (LiN(TMS)2) or lithium diisopropylamide (LDA). This metallation must be conducted under strictly controlled low-temperature conditions, typically ranging from -82°C to -40°C, to prevent side reactions and ensure the formation of the kinetic enolate. Once formed, this reactive nucleophile attacks the terminal position of the activated epoxide, such as 2(S)-glycidyl p-toluenesulfonate. This nucleophilic ring-opening event is highly regioselective, occurring exclusively at the less hindered terminus of the epoxide ring, which is crucial for establishing the correct connectivity for the HIV inhibitor backbone.
Controlling the stereochemistry at the newly formed asymmetric center is paramount for biological activity. The use of non-racemic, chirally pure glycidyl derivatives ensures that the resulting epoxide intermediate possesses the desired stereoconfiguration with high diastereoselectivity. This dual control—regioselectivity of the ring opening and stereoselectivity from the chiral starting material—eliminates the need for difficult downstream separations of unwanted isomers. From an impurity profile perspective, this mechanism minimizes the formation of double-addition byproducts, a common issue when reacting stabilized carbanions with epoxides. The result is a cleaner reaction mixture that simplifies purification, ultimately delivering a high-purity intermediate suitable for subsequent coupling steps without extensive chromatographic intervention.
How to Synthesize L-735,524 Intermediates Efficiently
Executing this synthesis requires careful attention to reaction parameters, particularly temperature control and reagent stoichiometry. The initial formation of the amide starting material involves acylation of aminoindanol followed by ketal protection, setting the stage for the key coupling event. The subsequent enolate formation and epoxide coupling are performed in ether solvents like tetrahydrofuran (THF) under an inert atmosphere. Following the coupling, the resulting epoxide intermediate is coupled with a chiral piperazine amine in a protic solvent like isopropanol at elevated temperatures. This robust sequence allows for the scalable production of the penultimate compound, which can then be deprotected to yield the final active pharmaceutical ingredient. Detailed standardized synthesis steps are provided in the guide below.
- Preparation of the chiral amide starting material via acylation of aminoindanol followed by ketal protection.
- Generation of the amide enolate using strong bases like LiN(TMS)2 or LDA at low temperatures (-50°C to -20°C).
- Nucleophilic attack of the enolate on activated glycidyl derivatives (e.g., glycidyl tosylate) to form the key epoxide intermediate.
- Coupling of the epoxide intermediate with a chiral piperazine amine followed by deprotection to yield the final inhibitor.
Commercial Advantages for Procurement and Supply Chain Teams
Adopting this novel synthetic route offers substantial strategic benefits for pharmaceutical manufacturing operations, addressing key pain points related to cost, safety, and scalability. By eliminating the reliance on osmium tetroxide and reducing the total number of synthetic steps, the process inherently lowers the cost of goods sold (COGS). The removal of toxic heavy metals simplifies waste management protocols and reduces the regulatory burden associated with residual metal testing in the final API. Furthermore, the use of common organic solvents and commercially available reagents enhances supply chain resilience, reducing the risk of production delays caused by specialty chemical shortages.
- Cost Reduction in Manufacturing: The streamlined nature of this synthesis directly impacts the bottom line by minimizing raw material consumption and processing time. Eliminating the 12-step legacy route in favor of a shorter sequence reduces labor costs, utility usage, and solvent waste disposal fees. The avoidance of expensive chiral auxiliaries and toxic oxidants further contributes to significant cost savings, making the production of HIV protease inhibitors more economically sustainable without compromising on quality or yield.
- Enhanced Supply Chain Reliability: The reagents required for this method, such as glycidyl tosylates and standard amide precursors, are widely available from multiple global suppliers. This diversification of the supply base mitigates the risk of single-source dependency. Additionally, the robustness of the reaction conditions allows for flexible manufacturing scheduling, ensuring that production targets can be met consistently. This reliability is critical for maintaining uninterrupted supply of life-saving antiretroviral medications to global markets.
- Scalability and Environmental Compliance: The process has been designed with commercial scale-up in mind, utilizing crystallization techniques for product isolation rather than complex chromatography. This facilitates the transition from kilogram to multi-ton production scales. Moreover, the reduction in hazardous waste generation aligns with increasingly stringent environmental regulations, positioning manufacturers as responsible stewards of sustainability. The ability to produce high-purity intermediates with a smaller environmental footprint is a key competitive advantage in the modern pharmaceutical landscape.
Frequently Asked Questions (FAQ)
The following questions address common technical and operational inquiries regarding the implementation of this synthetic methodology. These insights are derived directly from the experimental data and process descriptions found in the patent literature, providing clarity on feasibility and performance.
Q: How does this new method improve upon the conventional 12-step synthesis?
A: The conventional method requires 12 steps involving alkylated hydroxy-protected furanones and toxic osmium tetroxide. The novel route utilizes direct amide enolate alkylation with activated glycidol, significantly reducing step count and eliminating hazardous heavy metal oxidants.
Q: What ensures the stereochemical purity of the epoxide intermediate?
A: High diastereoselectivity is achieved by using non-racemic activated glycidyl derivatives, such as 2(S)-glycidyl p-toluenesulfonate, in combination with controlled low-temperature enolate formation, ensuring the correct configuration at the new asymmetric center.
Q: Is the piperazine coupling step scalable for industrial production?
A: Yes, the coupling reaction between the epoxide intermediate and the piperazine amine is performed in isopropanol at reflux temperatures, utilizing robust crystallization techniques for isolation, which supports large-scale commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable L-735,524 Intermediate Supplier
At NINGBO INNO PHARMCHEM, we understand the critical importance of efficient and scalable synthesis routes for complex pharmaceutical intermediates. Our team of expert process chemists has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project moves seamlessly from development to market. We are committed to delivering high-purity intermediates that meet stringent purity specifications, supported by our rigorous QC labs and state-of-the-art analytical capabilities. Our facility is equipped to handle the specific requirements of amide enolate chemistry and chiral epoxide coupling, guaranteeing consistent quality and supply continuity for your HIV inhibitor programs.
We invite you to collaborate with us to optimize your supply chain and reduce manufacturing costs. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our advanced manufacturing capabilities can support your commercial goals. Let us be your partner in bringing next-generation antiretroviral therapies to patients worldwide.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →