Advanced Synthesis of Nifuratel Related Substance C for Global Pharmaceutical Quality Control
Advanced Synthesis of Nifuratel Related Substance C for Global Pharmaceutical Quality Control
The pharmaceutical industry faces relentless pressure to ensure the safety and efficacy of active pharmaceutical ingredients (APIs), particularly regarding the control of genotoxic or harmful impurities. Patent CN112745273A introduces a groundbreaking preparation method for Nifuratel Related Substance C hydrochloride, a critical reference standard required for the rigorous quality control of the broad-spectrum antimicrobial agent Nifuratel. This patent details a robust synthetic route that transitions away from the traditional, inefficient isolation methods towards a direct, high-yield chemical synthesis. By leveraging a straightforward cyclization strategy starting from epichlorohydrin, this technology addresses the significant bottleneck of obtaining high-purity impurity standards, which are essential for validating analytical methods and ensuring patient safety in compliance with international pharmacopoeia standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the acquisition of Nifuratel Related Substance C has been a formidable challenge for quality control laboratories and research institutions worldwide. The conventional approach relied heavily on the isolation of this specific impurity directly from the crude reaction liquid of the main Nifuratel synthesis. This process typically necessitated the use of preparative liquid chromatography (Prep-HPLC), a technique that is inherently low-throughput, expensive, and difficult to scale. The yield obtained through such isolation methods was notoriously low, often resulting in insufficient quantities of the reference standard for comprehensive method validation or stability studies. Furthermore, the complexity of separating trace impurities from a matrix of structurally similar by-products often led to issues with purity, compromising the accuracy of quantitative analysis. These limitations severely restricted the ability of manufacturers to fully characterize the impurity profile of Nifuratel, posing a risk to regulatory compliance and drug safety.
The Novel Approach
In stark contrast to the arduous isolation techniques, the novel approach disclosed in the patent utilizes a dedicated synthetic pathway designed specifically for the target molecule. This method constructs the oxazolidinone core de novo using readily available starting materials, bypassing the need for complex separation from the parent drug's reaction mixture. The process involves a controlled ring-opening of epichlorohydrin followed by a precise cyclization with diethyl carbonate. This strategic shift allows for the production of the hydrochloride salt in substantial quantities with exceptional purity levels exceeding 99.9%. By decoupling the production of the impurity standard from the main API synthesis, manufacturers gain a reliable, independent source of high-quality reference material. This not only simplifies the supply chain for analytical standards but also ensures that the impurity profiles used for testing are consistent, well-characterized, and available in the volumes necessary for global regulatory submissions.
Mechanistic Insights into Base-Catalyzed Oxazolidinone Cyclization
The core of this innovative synthesis lies in the efficient construction of the 2-oxazolidinone ring system, a structural motif common in many bioactive molecules. The reaction mechanism initiates with the nucleophilic attack of hydrazine hydrate on the less hindered carbon of the epichlorohydrin epoxide ring. This ring-opening step generates a key intermediate, 3-chloro-2-hydroxy-propylhydrazine, which possesses both a nucleophilic amine and a hydroxyl group positioned perfectly for subsequent cyclization. In the second stage, the introduction of diethyl carbonate serves as a carbonyl source. Under the influence of an alkaline catalyst, such as sodium ethoxide or sodium hydroxide, the hydrazine nitrogen attacks the carbonyl carbon of the carbonate. This is followed by an intramolecular nucleophilic substitution where the hydroxyl group displaces the ethoxy leaving group, closing the five-membered ring to form the oxazolidinone structure. The careful control of temperature between 50-70°C and the stoichiometric balance of the base are critical to driving this equilibrium towards the desired cyclic product while minimizing side reactions such as polymerization or over-alkylation.
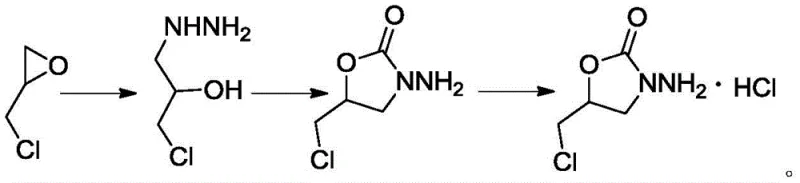
Furthermore, the mechanistic pathway includes a crucial purification strategy that leverages the physicochemical properties of the hydrochloride salt. Following the cyclization, the reaction mixture is treated with hydrochloric acid to adjust the pH, facilitating the precipitation of the product as its hydrochloride salt. This salt formation step is not merely a isolation technique but a powerful purification tool. The specific solubility profile of the hydrochloride salt in alcoholic solvents allows for effective removal of unreacted starting materials and organic by-products through a simple pulping process. This eliminates the need for chromatographic purification, which is often the most costly and time-consuming step in fine chemical synthesis. The result is a highly crystalline product with a defined melting point and spectral characteristics that match the theoretical structure, ensuring its suitability as a certified reference material for HPLC calibration and impurity tracking in Nifuratel batches.
How to Synthesize 3-Amino-5-Chloromethyl Oxazoline-2-Ketone Efficiently
The operational simplicity of this synthesis makes it an ideal candidate for technology transfer from the laboratory to pilot and commercial scales. The process is designed to be a one-pot or telescoped sequence that minimizes handling of intermediates, thereby reducing exposure risks and processing time. The initial step requires careful thermal management during the exothermic reaction between epichlorohydrin and hydrazine, followed by a distillation step to remove excess reagents. The subsequent cyclization is performed in common organic solvents like dichloromethane or ethanol, allowing for flexibility in solvent recovery and recycling. The final crystallization and pulping steps are standard unit operations found in any GMP facility, ensuring that the barrier to entry for manufacturing this high-value intermediate is low. For detailed standard operating procedures and specific parameter optimization, please refer to the technical guide below.
- React epichlorohydrin with hydrazine hydrate at 70-90°C to form 3-chloro-2-hydroxy-propylhydrazine.
- Cyclize the intermediate with diethyl carbonate and an alkaline catalyst at 50-70°C to form the oxazolidinone ring.
- Adjust pH, perform salt formation with concentrated hydrochloric acid, and purify via crystallization and pulping.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers transformative advantages in terms of cost structure and supply reliability. The shift from isolation to direct synthesis fundamentally changes the economics of producing Nifuratel Related Substance C. By utilizing commodity chemicals such as epichlorohydrin, hydrazine hydrate, and diethyl carbonate, the raw material costs are significantly reduced compared to the specialized solvents and columns required for preparative chromatography. Moreover, the elimination of complex purification steps drastically shortens the production cycle time, allowing for faster turnaround on orders for reference standards. This efficiency translates into a more resilient supply chain, capable of meeting the fluctuating demands of pharmaceutical quality control laboratories without the bottlenecks associated with low-yield isolation processes.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven by the high atom economy and the avoidance of expensive separation technologies. Traditional isolation methods suffer from poor mass balance, where the majority of the target compound is lost in the mother liquor or during column loading. In contrast, this direct synthesis achieves yields approaching 80%, effectively doubling the output per batch compared to legacy methods. Additionally, the use of simple filtration and pulping for purification removes the capital expenditure and operational costs associated with maintaining and running preparative HPLC systems. The reduction in solvent consumption and waste generation further contributes to a lower overall cost of goods sold, making the sourcing of this critical impurity standard more budget-friendly for pharmaceutical companies.
- Enhanced Supply Chain Reliability: Supply continuity is a paramount concern for global pharmaceutical operations, and this synthesis route mitigates many common risks. The raw materials employed are bulk chemicals produced by numerous suppliers worldwide, reducing the dependency on single-source vendors or niche intermediates. This diversification of the supply base ensures that production can continue even if one supplier faces disruptions. Furthermore, the robustness of the chemical process, characterized by wide operating windows for temperature and pH, means that batch failures due to minor process deviations are rare. This reliability ensures a steady stream of high-purity material, preventing delays in API release testing and regulatory filings that could otherwise arise from a shortage of qualified reference standards.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, the process is exceptionally well-suited for expansion. The reaction conditions are mild, avoiding the need for high-pressure equipment or cryogenic cooling, which simplifies the engineering requirements for scale-up. The waste stream is primarily composed of aqueous salts and recoverable alcohols, which are easier to treat and dispose of compared to the complex mixtures generated by chromatographic separations. This aligns with modern green chemistry principles and helps manufacturers meet increasingly stringent environmental regulations. The ability to scale from gram-scale laboratory synthesis to multi-kilogram commercial production without changing the fundamental chemistry provides a clear path for growing supply capacity in line with market demand for Nifuratel and its associated quality control materials.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Nifuratel Related Substance C. These insights are derived directly from the technical specifications and experimental data provided in the patent literature, ensuring that the information is grounded in verified scientific evidence. Understanding these aspects is crucial for R&D teams planning their analytical strategies and procurement teams evaluating potential suppliers for long-term contracts.
Q: Why is synthesizing Nifuratel Related Substance C directly preferred over isolation?
A: Direct synthesis avoids the low yields and high difficulty associated with isolating trace impurities from the main Nifuratel reaction liquid via preparative liquid chromatography, ensuring better availability for QC labs.
Q: What represents the critical quality attribute for this impurity standard?
A: High purity (>99.9%) is essential to ensure accurate quantification of impurities in the final API, meeting strict regulatory limits typically set below 0.10% for unknown impurities.
Q: Is this synthesis route scalable for industrial reference material production?
A: Yes, the process utilizes commodity raw materials like epichlorohydrin and diethyl carbonate and avoids complex chromatographic separations, making it highly suitable for large-scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Nifuratel Related Substance C Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality impurity standards play in the development and manufacturing of safe pharmaceutical products. Our team of expert chemists has thoroughly analyzed the synthetic route described in CN112745273A and is fully prepared to implement this advanced methodology for our clients. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements whether for early-stage method development or routine QC testing. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of Nifuratel Related Substance C we deliver meets the highest international standards for identity and assay.
We invite you to collaborate with us to optimize your supply chain for Nifuratel impurity controls. Our technical sales team is ready to provide a Customized Cost-Saving Analysis tailored to your specific consumption patterns and logistical needs. By partnering with us, you gain access to a stable, high-quality source of this critical reference material, along with comprehensive support documentation. Please contact our technical procurement team today to request specific COA data, route feasibility assessments, and competitive quotations that reflect the efficiencies of this next-generation synthesis platform.