Advanced Synthesis of Triazole Antifungal Intermediates for Commercial Scale Production
The global pharmaceutical landscape is witnessing a critical surge in demand for potent antifungal agents, driven by the rising incidence of invasive fungal infections caused by pathogens such as Candida, Aspergillus, and Cryptococcus. Patent CN100379733C introduces a groundbreaking methodology for the preparation of novel triazole derivatives, specifically targeting the structural backbone found in next-generation antifungal therapeutics. This intellectual property details a robust synthetic route that overcomes historical limitations in yield and stereoselectivity, providing a viable pathway for the mass production of high-purity active pharmaceutical ingredients. The core innovation lies in the strategic manipulation of substituents on the triazole and pyrimidine rings, which not only enhances biological activity but also streamlines the chemical synthesis. By leveraging specific nucleophilic addition reactions followed by selective reduction, this technology enables the efficient construction of complex chiral centers essential for drug efficacy. 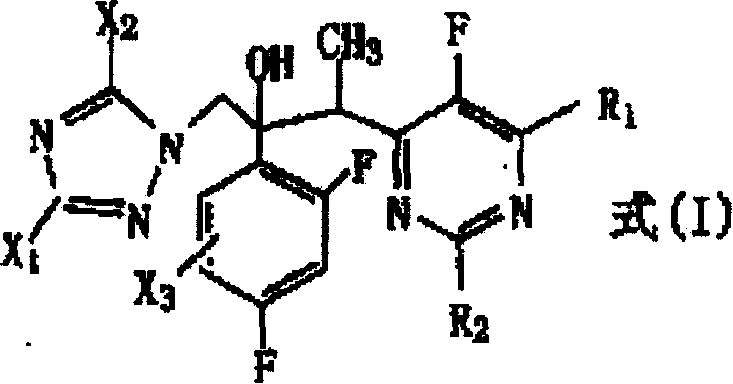
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of triazole antifungal intermediates has been plagued by inefficient reaction pathways that result in prohibitively low yields and poor stereochemical control. Prior art, such as that referenced in existing literature, often reports reaction yields of less than 5 percent for key nucleophilic addition steps, rendering these processes economically unfeasible for large-scale commercial manufacturing. Furthermore, conventional methods typically produce racemic mixtures or near-equimolar ratios of optical isomers, specifically a 1:1 ratio of (2R,3S/2S,3R) to (2R,3R/2S,3S) diastereomers. This lack of selectivity necessitates extensive and costly downstream purification processes to isolate the biologically active enantiomer, thereby inflating production costs and extending lead times. The reliance on non-optimized starting materials without specific ring substitutions further exacerbates these issues, leading to significant material waste and inconsistent batch quality that fails to meet the rigorous standards of modern regulatory bodies.
The Novel Approach
In stark contrast, the methodology disclosed in the patent utilizes specially substituted intermediates, specifically those with substituents at the 3-position or 5-position of the triazole ring, to dramatically alter the reaction kinetics and thermodynamics. This novel approach achieves reaction yields as high as 60 to 70 percent, representing a more than tenfold improvement over traditional literature methods. Crucially, the stereoselectivity is markedly enhanced, shifting the diastereomeric ratio from 1:1 to an impressive 5:1 in favor of the desired (2R,3S/2S,3R) configuration. This intrinsic selectivity reduces the burden on purification steps and maximizes the throughput of the active pharmaceutical ingredient. The process employs readily available precursors and operates under controlled conditions that are amenable to scale-up, ensuring a consistent supply of high-quality intermediates. 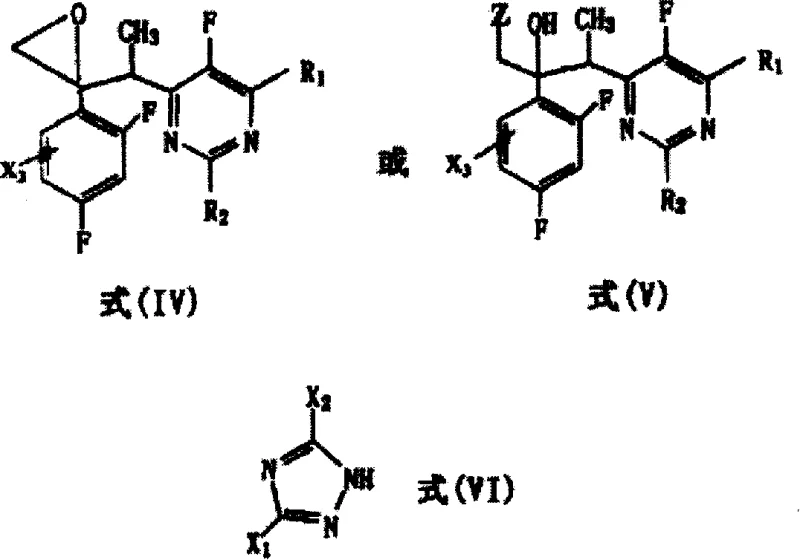
Mechanistic Insights into Nucleophilic Addition and Catalytic Reduction
The cornerstone of this synthesis is the nucleophilic addition reaction where a deprotonated pyrimidine derivative attacks a ketone substrate. In this critical step, a strong base such as lithium diisopropylamide (LDA) or sodium bis(trimethylsilyl)amide is employed to generate the 1'-deprotonated form of the pyrimidine compound in situ. This reaction is conducted in aprotic organic solvents like tetrahydrofuran or toluene under an inert atmosphere, strictly maintaining temperatures between -80°C and -10°C, with an optimal range of -70°C to -60°C. These cryogenic conditions are vital for controlling the reactivity of the anionic species and preventing side reactions that could degrade the sensitive heterocyclic rings. The resulting alkoxide intermediate is then quenched and processed to yield the hydroxy-substituted Formula (IA) compound, which serves as the pivotal scaffold for subsequent functionalization.
Following the formation of the carbon-carbon bond, the process involves a sophisticated reduction step to refine the molecular structure. The patent describes the conversion of Formula (IA) to Formula (IB) through catalytic hydrogenolysis or transfer catalytic hydrogenolysis. This step effectively removes specific halogen substituents, such as chlorine or bromine, from the triazole or pyrimidine rings without affecting other sensitive functional groups. The use of palladium-carbon catalysts, either under hydrogen pressure or with ammonium formate as a hydrogen donor, ensures a clean and selective transformation. 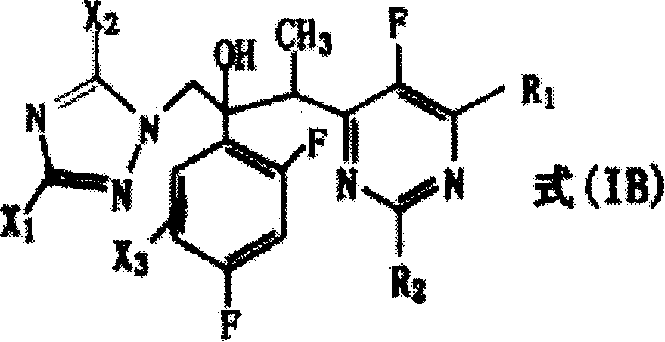 This mechanistic precision allows for the fine-tuning of the final drug candidate's pharmacokinetic properties, ensuring that the resulting molecule possesses the optimal balance of lipophilicity and metabolic stability required for effective antifungal activity in clinical settings.
This mechanistic precision allows for the fine-tuning of the final drug candidate's pharmacokinetic properties, ensuring that the resulting molecule possesses the optimal balance of lipophilicity and metabolic stability required for effective antifungal activity in clinical settings.
How to Synthesize Triazole Antifungal Intermediates Efficiently
To implement this advanced synthesis in a production environment, precise adherence to the reaction parameters outlined in the patent is essential for maximizing yield and purity. The process begins with the careful preparation of the deprotonated nucleophile, followed by the controlled addition of the electrophilic ketone, and concludes with a selective reduction and resolution sequence. Operators must maintain strict temperature control and inert atmospheric conditions throughout the nucleophilic addition phase to prevent degradation. The following guide outlines the standardized operational procedure derived from the patent examples, ensuring reproducibility and safety during scale-up operations. Detailed standard operating procedures for each unit operation are provided below to facilitate immediate technology transfer.
- Deprotonate the pyrimidine derivative (Formula II) using a strong base like LDA or NaHMDS in THF at low temperatures (-70°C to -60°C).
- React the resulting salt in situ with the ketone derivative (Formula III) to form the alcohol intermediate (Formula IA) with high stereoselectivity.
- Perform catalytic hydrogenolysis using Pd/C and hydrogen or ammonium formate to remove halogen substituents, yielding the final Formula (IB) compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route offers transformative economic benefits that directly impact the bottom line. The dramatic increase in reaction yield from single digits to over 60 percent fundamentally alters the cost structure of raw material consumption, significantly reducing the cost of goods sold per kilogram of finished product. By minimizing the generation of unwanted isomers and byproducts, the process lowers the load on waste treatment facilities and reduces the volume of solvents required for purification, contributing to substantial operational expenditure savings. Furthermore, the use of common industrial reagents like LDA and palladium on carbon ensures that the supply chain remains resilient against shortages of exotic or highly specialized catalysts. This reliability is crucial for maintaining continuous production schedules and meeting the demanding delivery timelines of global pharmaceutical clients.
- Cost Reduction in Manufacturing: The elimination of low-yield steps and the enhancement of stereoselectivity mean that less starting material is wasted, directly translating to lower raw material costs. The process avoids the need for complex chiral chromatography by leveraging diastereomeric salt formation with camphorsulfonic acid, a cost-effective resolution technique. Additionally, the ability to use transfer hydrogenolysis with ammonium formate provides a safer and potentially cheaper alternative to high-pressure hydrogenation equipment, reducing capital expenditure requirements for facility upgrades.
- Enhanced Supply Chain Reliability: The starting materials, including substituted pyrimidines and triazoles, are based on established chemical scaffolds with robust global supply chains. The synthesis does not rely on unstable intermediates that require immediate consumption, allowing for the stocking of key precursors to buffer against market volatility. The scalability of the reaction, explicitly noted in the patent as suitable for large-scale preparation, ensures that suppliers can ramp up production volumes rapidly to meet surges in demand without compromising quality or extending lead times.
- Scalability and Environmental Compliance: The process is designed with green chemistry principles in mind, utilizing efficient atom economy in the key coupling step. The reduction in solvent usage and waste generation simplifies environmental compliance and lowers the costs associated with hazardous waste disposal. The mild conditions for the resolution step and the flexibility in reduction methods allow the process to be adapted to various manufacturing sites, ensuring consistent quality across different production batches and facilitating global regulatory approval.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this triazole synthesis technology. These answers are derived directly from the experimental data and claims within the patent documentation, providing clarity on process capabilities and product specifications. Understanding these details is essential for R&D teams evaluating the feasibility of this route for their specific pipeline candidates. The responses cover aspects ranging from reaction conditions to final product isolation strategies.
Q: What is the primary advantage of this synthesis method over conventional literature methods?
A: The patented method significantly improves reaction yield from less than 5% in literature to 60-70%, while enhancing stereoselectivity ratios from 1:1 to 5:1, making it viable for industrial production.
Q: Which reducing agents are suitable for the dehalogenation step?
A: The process supports both catalytic hydrogenolysis using palladium-carbon catalysts under hydrogen pressure and transfer hydrogenolysis using ammonium formate, offering flexibility in equipment requirements.
Q: How is optical purity achieved in the final product?
A: Optical purity is secured through resolution using optically active acids such as 1S-(+) or 1R-(-)-10-camphorsulfonic acid, followed by fractional crystallization of the diastereomeric salts.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Triazole Antifungal Intermediate Supplier
At NINGBO INNO PHARMCHEM, we possess the technical expertise and infrastructure to translate this patented laboratory methodology into a robust commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with unwavering consistency. We operate stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify every batch against the highest industry standards. Our commitment to quality assurance means that every gram of triazole antifungal intermediate we deliver is backed by comprehensive documentation and full traceability.
We invite you to collaborate with us to optimize your supply chain and reduce your manufacturing costs. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our advanced synthesis capabilities can accelerate your drug development timeline and enhance your market competitiveness.