Advanced Purification Technology for Repaglinide: Enhancing Purity and Commercial Scalability
Introduction to Advanced Repaglinide Purification Technologies
The global demand for high-quality oral hypoglycemic agents continues to surge, driven by the increasing prevalence of Type II diabetes worldwide. At the forefront of this therapeutic class is Repaglinide, a non-sulfonylurea insulin secretagogue that offers rapid onset and short duration of action, mimicking physiological insulin secretion. However, the commercial viability of this critical Active Pharmaceutical Ingredient (API) has historically been constrained by significant challenges in achieving consistent high purity. Patent CN102702138B introduces a groundbreaking refinement methodology that fundamentally addresses these purity bottlenecks. By integrating a sophisticated multi-step purification protocol involving selective adsorption and targeted alkali hydrolysis, this technology transforms crude Repaglinide, often containing substantial impurities, into a pharmaceutical-grade substance with purity exceeding 99.6%. For R&D directors and procurement strategists, understanding the mechanistic nuances of this patent is essential for securing a reliable repaglinide supplier capable of delivering superior quality while optimizing supply chain economics.
The significance of this technological breakthrough extends beyond mere compliance with pharmacopoeial standards; it represents a paradigm shift in how complex organic molecules are refined post-synthesis. Traditional manufacturing routes often leave behind stubborn impurities, such as residual solvents, heavy metals, and structurally similar byproducts, which necessitate costly and yield-depleting purification cycles. The method disclosed in CN102702138B elegantly circumvents these issues by leveraging the differential solubility and chemical reactivity of impurities. Through a carefully orchestrated sequence of dissolution, adsorptive cleaning, chemical conversion of impurities, and precision recrystallization, the process ensures that the final product not only meets stringent safety profiles but also exhibits enhanced physical properties, such as improved powder flowability and dissolution rates, which are critical for downstream formulation.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art synthesis and purification strategies for Repaglinide have long been plagued by inherent inefficiencies that compromise both economic viability and product quality. For instance, early synthetic routes described in patents like US5312924 relied heavily on coupling agents such as N,N'-dicyclohexylcarbodiimide (DCC). While effective for bond formation, this approach generates N,N'-dicyclohexylurea (DCU) as a stoichiometric byproduct, which is notoriously difficult to remove completely without multiple, yield-reducing recrystallization steps. Furthermore, alternative methods utilizing carbonyldiimidazole (CDI) often result in mediocre yields ranging from merely 50% to 55%, alongside the burden of expensive reagent costs. Other approaches involving triphenylphosphine and azodicarboxylates, while improving yield, frequently suffer from low product purity due to the formation of phosphine oxide byproducts and other side reactions. These conventional limitations create a precarious supply chain environment where manufacturers struggle to balance cost against quality, often resulting in batch failures or the need for extensive, resource-intensive post-processing.
The Novel Approach
In stark contrast to these legacy methods, the novel approach detailed in CN102702138B offers a robust, four-step purification strategy that decouples purity enhancement from yield loss. Instead of relying solely on physical separation, this method actively converts specific impurities back into the desired product. The process begins with a selective dissolution in water-immiscible organic solvents to eliminate insoluble inorganic contaminants, followed by a critical adsorption step using materials like activated carbon or molecular sieves to sequester lipophilic impurities and bacterial endotoxins. The true innovation lies in the subsequent treatment with alkali metal alkoxides, which hydrolyzes residual ester protecting groups—common impurities in Repaglinide synthesis—back into the active acid form. This chemical "rescue" of the product significantly boosts the effective yield while simultaneously simplifying the impurity profile, paving the way for a final recrystallization step that delivers white crystals of exceptional purity and stability.
Mechanistic Insights into Adsorption and Alkali-Mediated Purification
The core of this purification technology rests on a deep understanding of intermolecular interactions and chemical kinetics. In the initial stages, the process exploits the solubility differences between the target molecule and various contaminant classes. Lipophilic impurities, often introduced during synthesis or from raw materials, are effectively captured by high-surface-area adsorbents. Specifically, the use of molecular sieves, such as Type A or X-type zeolites, provides a highly selective porous framework that traps molecules based on size and polarity. The crystalline structure of these aluminosilicates, characterized by a uniform network of pores and channels, allows for the precise exclusion of larger Repaglinide molecules while retaining smaller or more polar impurity species within the lattice. This selective adsorption is crucial for removing trace metals and organic residues that could otherwise catalyze degradation or trigger adverse biological responses.
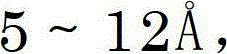
Following adsorption, the process employs a chemically transformative step that distinguishes it from standard physical purification. The addition of aqueous alkali metal alkoxides, such as sodium ethylate or potassium methylate, initiates a saponification-like reaction under controlled thermal conditions (typically 50-60°C). This step targets residual ester impurities, which are essentially protected forms of the Repaglinide carboxyl group that failed to deprotect in previous synthesis stages. Under basic conditions, these esters undergo hydrolysis, cleaving the protecting group and regenerating the free carboxylic acid of Repaglinide. This mechanism not only eliminates a major class of impurities but effectively recovers lost product, thereby enhancing the overall mass balance of the operation. The subsequent phase separation removes water-soluble salts and hydrolyzed byproducts, leaving an organic phase enriched with the target compound, ready for the final crystallization.
The final stage involves a meticulously controlled recrystallization driven by pH adjustment and temperature modulation. By introducing an alcoholic solvent and adjusting the pH to a narrow acidic window of 3.0 to 5.5, the solubility of Repaglinide is precisely manipulated to induce nucleation and crystal growth. This pH range is critical; it ensures that the carboxylic acid group is protonated sufficiently to reduce solubility without causing precipitation of acidic impurities or degradation of the molecule. Slow cooling from elevated temperatures (60-70°C) down to ambient or lower temperatures allows for the formation of large, well-defined crystals with minimal solvent inclusion. This results in a product with superior physical characteristics, including improved flowability and intrinsic dissolution rate, which are vital parameters for the manufacturability of the final dosage form.
How to Synthesize High-Purity Repaglinide Efficiently
Implementing this purification protocol requires strict adherence to the operational parameters defined in the patent to ensure reproducibility and optimal outcomes. The process is designed to be compatible with existing crude Repaglinide obtained from various synthetic routes, making it a versatile upgrade for current manufacturing lines. Operators must carefully manage the ratio of adsorbent to solution volume, typically maintaining a range of 0.1% to 1% (g/ml), and control the contact time and temperature to maximize impurity uptake without sacrificing product retention. Similarly, the alkali treatment phase demands precise monitoring of reaction time and temperature to ensure complete hydrolysis of esters while preventing base-catalyzed degradation of the sensitive amide backbone. The following guide outlines the standardized steps for executing this advanced purification strategy.
- Dissolve crude Repaglinide in a water-immiscible organic solvent (e.g., chloroform, ethyl acetate) and filter to remove insoluble mineral impurities.
- Treat the filtrate with adsorptive inorganic substances such as activated carbon, aluminum oxide, or molecular sieves to remove lipophilic impurities and endotoxins.
- Process the secondary filtrate with an aqueous solution of alkali metal alkoxides (e.g., sodium ethylate) at 50-60°C to hydrolyze residual ester impurities back into Repaglinide.
- Add alcoholic solvent, adjust pH to 3.0-5.5 using acid, and perform controlled temperature recrystallization to isolate high-purity white crystals.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this purification technology translates into tangible strategic advantages that extend far beyond simple regulatory compliance. The primary value proposition lies in the substantial optimization of the cost structure associated with API manufacturing. By recovering product from what would traditionally be classified as waste (ester impurities), the process effectively increases the yield per batch without requiring additional raw material inputs. This yield enhancement directly correlates to a reduction in the cost of goods sold (COGS), allowing for more competitive pricing models in the global marketplace. Furthermore, the elimination of complex chromatographic separations and the reduction in the number of recrystallization cycles significantly lower solvent consumption and energy usage, contributing to a leaner and more sustainable production footprint.
- Cost Reduction in Manufacturing: The economic impact of this technology is profound, primarily driven by the conversion of impurities into saleable product. In conventional processes, ester impurities represent a direct loss of yield, requiring expensive disposal or complex reprocessing. By hydrolyzing these esters back into Repaglinide, the process maximizes the utility of every kilogram of crude input. Additionally, the use of inexpensive, regenerable adsorbents like activated carbon and molecular sieves replaces costly specialty resins or extensive column chromatography, drastically reducing consumable expenses. The streamlined workflow also minimizes labor hours and equipment occupancy time, further driving down overhead costs and enhancing overall operational efficiency.
- Enhanced Supply Chain Reliability: Supply continuity is a critical concern for pharmaceutical buyers, and this robust purification method significantly mitigates the risk of batch failures. The process is less sensitive to variations in crude quality compared to traditional methods, as the adsorption and hydrolysis steps act as powerful buffers against impurity spikes. This resilience ensures a more consistent output of high-purity material, reducing the likelihood of out-of-specification (OOS) results that can disrupt supply schedules. Moreover, the reliance on common, commercially available reagents and solvents reduces dependency on niche suppliers, safeguarding the production line against raw material shortages and price volatility in the upstream chemical market.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, the process is exceptionally well-suited for commercial scale-up of complex pharmaceutical intermediates. The unit operations involved—filtration, stirring, heating, and crystallization—are standard in the chemical industry and scale linearly from pilot plant to multi-ton production without significant engineering hurdles. The reduction in solvent waste and the avoidance of heavy metal catalysts or hazardous coupling reagents align with increasingly stringent global environmental regulations. This "green chemistry" aspect not only simplifies waste disposal logistics but also enhances the corporate sustainability profile of the supply chain, a factor of growing importance to end-user pharmaceutical companies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation and benefits of this purification technology. These insights are derived directly from the experimental data and theoretical framework presented in the patent documentation, providing a clear understanding of how this method resolves historical manufacturing pain points. Understanding these details is crucial for technical teams evaluating the feasibility of integrating this process into their existing production workflows or for procurement specialists assessing the long-term value of sourcing from manufacturers utilizing this advanced methodology.
Q: How does this purification method overcome the limitations of traditional Repaglinide synthesis?
A: Traditional methods often suffer from difficult-to-remove byproducts like N,N'-dicyclohexylurea (DCU) or incomplete deprotection of carboxyl groups. This novel method utilizes a unique combination of adsorptive filtration and alkali hydrolysis to convert residual ester impurities back into the target molecule, simultaneously boosting yield and purity to over 99.6%.
Q: What are the primary drivers for cost reduction in this manufacturing process?
A: Cost efficiency is achieved through multiple mechanisms: the recovery of target product from ester impurities via hydrolysis effectively increases overall yield; the use of common adsorbents like molecular sieves replaces expensive chromatographic steps; and the simplified workflow reduces solvent consumption and processing time compared to repeated recrystallizations required by older techniques.
Q: Is this purification technology suitable for large-scale industrial production?
A: Yes, the process is explicitly designed for industrial scalability. It relies on standard unit operations such as agitation, filtration, phase separation, and crystallization, avoiding complex or hazardous reactions. The robust control parameters, including specific pH ranges and temperature windows, ensure consistent quality and supply continuity for commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Repaglinide Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory innovation to commercial reality requires more than just a patent; it demands deep process engineering expertise and a commitment to uncompromising quality. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of advanced purification technologies like CN102702138B are fully realized in large-scale manufacturing. Our facilities are equipped with state-of-the-art rigorous QC labs and analytical instrumentation capable of verifying stringent purity specifications, guaranteeing that every batch of Repaglinide meets the highest global pharmacopoeial standards. We understand the critical nature of supply chain stability for life-saving medications and are dedicated to providing a seamless, reliable source of high-quality API intermediates.
We invite forward-thinking pharmaceutical companies to collaborate with us to optimize their supply chains and reduce manufacturing costs. By leveraging our technical capabilities, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. Our technical procurement team is ready to provide specific COA data and route feasibility assessments to demonstrate how our advanced purification processes can enhance your product portfolio. Contact us today to discuss how we can support your development goals with reliable, cost-effective, and high-purity Repaglinide solutions.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →