Optimizing L-Erythro-Biopterin Production: A Mercaptan-Free Strategy for Commercial Scale-Up
The pharmaceutical landscape for treating atypical hyperphenylalaninemia relies heavily on the availability of high-purity intermediates such as L-erythro-biopterin, a critical precursor to Sapropterin hydrochloride. Patent CN101792446A introduces a transformative preparation method that addresses longstanding inefficiencies in the synthesis of this vital pteridine derivative. By shifting away from malodorous thiol-based degradation pathways, this technology offers a cleaner, more operationally friendly route starting from L-(+)-arabinose. For R&D directors and procurement specialists, understanding this shift is crucial, as it represents a move towards greener chemistry without compromising the structural integrity required for downstream hydrogenation into the active pharmaceutical ingredient. This report analyzes the technical merits and commercial implications of adopting this mercaptan-free synthesis strategy.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial preparation of 5-deoxy-L-arabinose, the sugar backbone essential for biopterin synthesis, has relied heavily on the degradation of L-rhamnose. As illustrated in the prior art reaction schemes, this conventional pathway necessitates the use of ethanethiol to form dithioacetals, which are subsequently oxidized to sulfones before undergoing alkaline elimination to shorten the carbon chain. 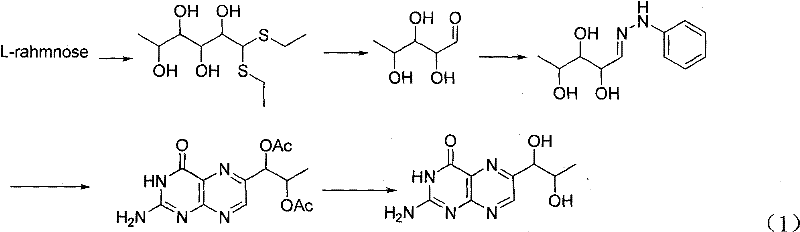 This reliance on volatile sulfur compounds presents severe logistical and environmental challenges, including the absolute necessity for specialized deodorization equipment to manage the unbearable stench associated with mercaptans. Furthermore, the multi-step nature of converting rhamnose to the key arabinose intermediate often results in prolonged production cycles and lower overall throughput, creating bottlenecks for supply chain managers seeking consistent volume. The handling of low-boiling thiols also introduces significant safety hazards and regulatory compliance burdens regarding atmospheric emissions, making the traditional route increasingly untenable for modern, eco-conscious manufacturing facilities.
This reliance on volatile sulfur compounds presents severe logistical and environmental challenges, including the absolute necessity for specialized deodorization equipment to manage the unbearable stench associated with mercaptans. Furthermore, the multi-step nature of converting rhamnose to the key arabinose intermediate often results in prolonged production cycles and lower overall throughput, creating bottlenecks for supply chain managers seeking consistent volume. The handling of low-boiling thiols also introduces significant safety hazards and regulatory compliance burdens regarding atmospheric emissions, making the traditional route increasingly untenable for modern, eco-conscious manufacturing facilities.
The Novel Approach
In stark contrast, the methodology disclosed in CN101792446A circumvents these issues by utilizing a direct functionalization strategy on L-(+)-arabinose. Instead of degrading a higher sugar, the process protects the sugar via glycosidation and then employs sulfonyl chlorides, such as p-toluenesulfonyl chloride or methanesulfonyl chloride, to activate specific hydroxyl groups. This activation allows for a clean reductive removal of the sulfonate ester using hydride sources like lithium aluminum hydride or sodium borohydride, effectively achieving the desired deoxygenation without generating foul-smelling byproducts. The subsequent steps involve hydrazone formation, acylation, and a sophisticated condensation with 2,4,5-triaminopyrimidinone followed by oxidation to close the pteridine ring. 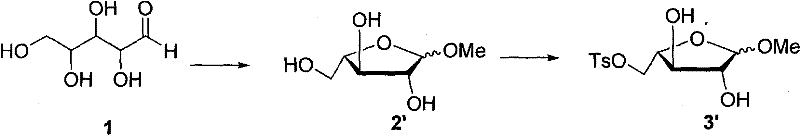 This approach not only eliminates the need for odor control infrastructure but also streamlines the workflow, offering milder reaction conditions that are easier to control on a large scale, thereby enhancing the feasibility of commercial scale-up of complex pharmaceutical intermediates.
This approach not only eliminates the need for odor control infrastructure but also streamlines the workflow, offering milder reaction conditions that are easier to control on a large scale, thereby enhancing the feasibility of commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Sulfonyl-Mediated Deoxygenation and Pteridine Formation
The core innovation of this synthesis lies in the mechanistic elegance of the sulfonylation-reduction sequence, which replaces the harsh thiol-oxidation-elimination triad. In the initial stages, L-(+)-arabinose undergoes glycosidation with an alcohol (C1-C10 chain) under acidic catalysis to form a stable glycoside, protecting the anomeric center. The subsequent introduction of a sulfonyl group converts a primary or secondary hydroxyl into an excellent leaving group. When treated with a strong reducing agent like LiAlH4, the carbon-sulfur bond is cleaved reductively, yielding the deoxygenated sugar scaffold with high stereochemical fidelity. This mechanism avoids the radical pathways often associated with thiol chemistry, leading to a cleaner impurity profile that is highly attractive for R&D teams focused on purity specifications. The absence of sulfur-containing side products simplifies downstream purification, reducing the load on chromatography or crystallization steps.
Following the construction of the chiral side chain, the formation of the pteridine core involves a condensation reaction between the acylated hydrazone derivative and 2,4,5-triaminopyrimidinone. This cyclization is driven by the nucleophilic attack of the aminopyrimidine on the hydrazone carbon, followed by an oxidative aromatization step using agents like iodine or hydrogen peroxide. The choice of oxidant is critical; iodine provides a controlled oxidation potential that minimizes over-oxidation of the sensitive dihydroxypropyl side chain, ensuring the final product retains the specific (1R,2S) configuration required for biological activity. This precise control over the oxidation state is a key factor in achieving the high purity necessary for clinical applications, demonstrating how mechanistic understanding directly translates to product quality in high-purity pharmaceutical intermediate manufacturing.
How to Synthesize L-Erythro-Biopterin Efficiently
The synthesis protocol outlined in the patent provides a robust framework for producing L-erythro-biopterin with improved operational safety and efficiency. The process begins with the protection of L-(+)-arabinose, followed by activation and reduction to establish the correct carbon skeleton. Subsequent functionalization with phenylhydrazine and acyl groups prepares the molecule for the critical ring-closing step with the pyrimidine precursor. The detailed standardized synthesis steps below outline the specific reagents, stoichiometry, and conditions required to replicate this high-yielding pathway in a pilot or production setting.
- Perform glycosidation of L-(+)-arabinose with alcohol followed by sulfonylation using sulfonyl chloride.
- Execute reductive removal of the sulfonate group and deprotect the acetal to yield 5-deoxy-L-arabinose.
- React the resulting sugar with phenylhydrazine, acylate, condense with 2,4,5-triaminopyrimidinone, oxidize, and finally deacylate.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this mercaptan-free synthesis route offers tangible benefits that extend beyond simple chemical yield. The most immediate impact is the elimination of capital expenditure related to odor scrubbing and deodorization systems, which are mandatory when handling ethanethiol. This reduction in infrastructure requirements directly lowers the barrier to entry for manufacturing this intermediate, allowing for more flexible production scheduling and potentially lower overhead costs per kilogram. Additionally, the use of solid or high-boiling liquid reagents like sulfonyl chlorides improves workplace safety and reduces the risk of volatile organic compound (VOC) emissions, aligning with increasingly stringent environmental regulations globally.
- Cost Reduction in Manufacturing: The removal of thiol-based reagents eliminates the need for expensive waste treatment processes specifically designed to neutralize sulfur odors, leading to substantial cost savings in utility and waste management. Furthermore, the milder reaction conditions reduce energy consumption associated with heating and cooling cycles, while the simplified workup procedures minimize solvent usage and processing time. By avoiding the complex degradation of L-rhamnose, the raw material costs can also be optimized, as L-arabinose is a readily available starting material that does not require the extensive preprocessing seen in older methods.
- Enhanced Supply Chain Reliability: The streamlined nature of this synthesis reduces the number of unit operations required to reach the key intermediate, thereby decreasing the probability of batch failures and production delays. The stability of the intermediates formed during the sulfonylation and reduction steps ensures that the process is robust against minor fluctuations in temperature or mixing rates, which is critical for maintaining consistent supply to downstream API manufacturers. This reliability is further bolstered by the use of common, commercially available reagents that are less subject to supply chain disruptions compared to specialized sulfur compounds.
- Scalability and Environmental Compliance: The absence of noxious gases makes this process inherently more scalable, as it does not require the engineering controls necessary to contain toxic vapors in large reactors. This facilitates easier technology transfer from laboratory to plant scale, reducing the lead time for high-purity pharmaceutical intermediates to reach the market. Moreover, the reduced environmental footprint supports corporate sustainability goals, making the supply chain more resilient to future regulatory changes regarding chemical emissions and worker safety standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis route. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on how this method compares to established industry standards. Understanding these nuances is essential for stakeholders evaluating the feasibility of integrating this technology into their existing production portfolios.
Q: Why is the sulfonylation route preferred over the traditional thiol method for L-erythro-biopterin?
A: The traditional method utilizes ethanethiol, which has an unbearable stench and requires specialized deodorization equipment. The novel sulfonylation route eliminates mercaptans entirely, optimizing the working environment and reducing infrastructure costs.
Q: What are the key reaction conditions for the condensation step in this synthesis?
A: The condensation of the ester compound with 2,4,5-triaminopyrimidinone occurs at temperatures between 0°C and 100°C, typically followed by oxidation using iodine or peroxides to form the pteridine ring system.
Q: How does this process impact the production cycle time?
A: By avoiding the complex multi-step degradation of L-rhamnose via sulfones and eliminating the need for odor control processing, the new method significantly shortens the overall production cycle and simplifies operational procedures.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable L-Erythro-Biopterin Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that efficient intermediate synthesis plays in the global supply of life-saving medications like Sapropterin hydrochloride. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from patent literature to industrial reality is seamless and compliant. We are committed to delivering L-erythro-biopterin with stringent purity specifications, supported by our rigorous QC labs that monitor every step of the sulfonylation and condensation process to guarantee batch-to-batch consistency.
We invite you to engage with our technical procurement team to discuss how this advanced manufacturing route can optimize your supply chain dynamics. By requesting a Customized Cost-Saving Analysis, you can gain deeper visibility into the potential economic benefits of switching to this mercaptan-free method. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your project's unique requirements, ensuring a partnership built on transparency and technical excellence.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →