Advanced Synthesis of Lenalidomide Key Intermediate for Commercial Scale-up
Advanced Synthesis of Lenalidomide Key Intermediate for Commercial Scale-up
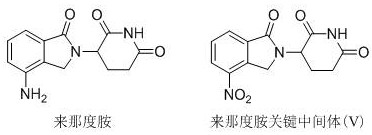
The pharmaceutical industry continuously seeks robust and scalable pathways for producing critical oncology therapeutics, and the synthesis of lenalidomide remains a focal point for process chemists worldwide. Patent CN111548341B introduces a transformative chemical synthesis method for the key intermediate 3-(4-nitro-1-oxo-1,3-dihydro-2H-isoindol-2-yl)piperidine-2,6-dione, addressing long-standing challenges in yield and operational safety. This innovation utilizes 3-nitrophthalic acid as a starting material, proceeding through cyclic amination, carbonyl reduction, condensation, and selective oxidation to deliver the target molecule with exceptional efficiency. By streamlining the reaction sequence and employing mild conditions, this technology offers a viable alternative to traditional routes that often suffer from harsh reagents and complex purification burdens. For R&D directors and procurement specialists, understanding the nuances of this patent is essential for securing a reliable pharmaceutical intermediates supplier capable of meeting stringent quality demands. The structural relationship between the final drug and this critical precursor is fundamental to the efficacy of the treatment regimen.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the production of lenalidomide precursors has been plagued by inefficient synthetic routes that impose significant burdens on manufacturing infrastructure and cost structures. Prior art methods, such as those disclosed in US 5656325517, often require multi-step sequences involving long-time reflux reactions that degrade product purity and complicate separation processes. Other approaches, like those in CN 101580501, rely on sodium amide as a catalyst, a reagent that is notoriously difficult to prepare and store safely, thereby limiting the effectiveness of large-scale preparation. Furthermore, methods utilizing mixed acid for nitration or expensive catalysts like 5-trimethyl ammonium o-iodoxybenzoic acid inner salt generate substantial three wastes that are difficult to control and require specialized equipment to handle safely. These conventional pathways often result in low product purity, making the downstream purification process both time-consuming and economically draining for any fine chemical manufacturer attempting to commercialize the drug.
The Novel Approach
In stark contrast to the cumbersome legacy processes, the novel approach defined in patent CN111548341B leverages a streamlined four-step sequence that prioritizes operational simplicity and environmental compatibility. By initiating the synthesis with 3-nitrophthalic acid, a readily available and cost-effective raw material, the method eliminates the need for hazardous or hard-to-source reagents found in older patents. The reaction conditions are notably mild, operating at moderate temperatures that reduce energy consumption and lower the requirements for specialized high-pressure or high-temperature equipment. This new route ensures that product separation and purification are straightforward, typically involving standard extraction and pulping techniques rather than complex chromatographic methods. Consequently, this approach not only enhances the overall yield but also significantly improves the safety profile of the manufacturing process, making it an ideal candidate for cost reduction in pharmaceutical intermediates manufacturing where efficiency and compliance are paramount.
Mechanistic Insights into Cu-Catalyzed Selective Oxidation and Reduction
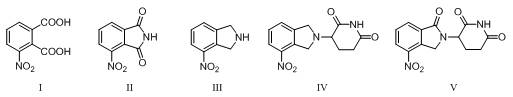
The core of this synthetic breakthrough lies in the precise control of chemical transformations, particularly during the carbonyl reduction and selective oxidation stages. The process begins with the cyclization ammoniation of 3-nitrophthalic acid with urea in glacial acetic acid at 115°C, forming 3-nitrophthalimide with high conversion rates. Subsequently, the carbonyl reduction is executed using a system of sodium borohydride and boron trifluoride diethyl etherate in anhydrous tetrahydrofuran, where the molar ratios are critically optimized to prevent incomplete substrate conversion or excessive reagent costs. The reaction temperature is carefully maintained between 25°C and 66°C to balance reaction rate and selectivity, ensuring the formation of 4-nitroisoindoline without generating unwanted by-products. This meticulous attention to stoichiometry and thermal conditions underscores the robustness of the method, providing R&D teams with a reproducible protocol that minimizes batch-to-batch variability and ensures consistent quality for high-purity pharmaceutical intermediates.
Following the reduction, the condensation with 3-bromo-2,6-piperidinedione and the subsequent selective oxidation represent the most critical steps for establishing the final molecular architecture. The oxidation step employs a copper salt catalyst, such as copper sulfate pentahydrate or cuprous bromide, in the presence of an oxidant like tert-butyl hydroperoxide under an oxygen atmosphere. This catalytic system facilitates the conversion of the dihydro-isoindol derivative to the target oxo-isoindol structure with remarkable specificity. The reaction temperature is controlled between 50°C and 90°C to prevent excessive oxidation by-products while ensuring complete substrate conversion. The use of additives like N-hydroxyphthalimides further refines the reaction environment, enhancing the yield and purity of the final product. This mechanistic precision allows for the commercial scale-up of complex pharmaceutical intermediates with confidence, as the process is designed to mitigate impurity formation and simplify the final isolation steps.
How to Synthesize 3-(4-nitro-1-oxo-1,3-dihydro-2H-isoindol-2-yl)piperidine-2,6-dione Efficiently
Implementing this synthesis route requires a systematic approach to reaction management and post-treatment protocols to maximize efficiency and yield. The process is designed to be operationally simple, allowing technical teams to execute the synthesis with standard laboratory or plant equipment without the need for exotic infrastructure. Detailed standard operating procedures focus on the precise addition of reagents, such as the dropwise addition of boron trifluoride diethyl etherate at controlled temperatures to manage exothermic risks. Post-reaction treatments involve quenching with saturated sodium bicarbonate, pH adjustments for phase separation, and pulping with solvent mixtures like n-hexane and ethyl acetate to isolate the solid product. These steps are critical for removing residual reagents and by-products, ensuring the final material meets stringent purity specifications required for downstream drug synthesis. For a comprehensive guide on the exact parameters and safety measures, refer to the standardized synthesis steps provided below.
- Cyclization ammoniation of 3-nitrophthalic acid with urea in glacial acetic acid at 115°C to form 3-nitrophthalimide.
- Carbonyl reduction using sodium borohydride and boron trifluoride diethyl etherate in tetrahydrofuran to yield 4-nitroisoindoline.
- Condensation with 3-bromo-2,6-piperidinedione in acetonitrile followed by selective copper-catalyzed oxidation to finalize the intermediate.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic procurement perspective, this synthesis method offers substantial cost savings and supply chain reliability by fundamentally altering the input material landscape and processing requirements. The reliance on 3-nitrophthalic acid, a commodity chemical, eliminates the dependency on specialized or imported starting materials that often cause supply bottlenecks and price volatility. Furthermore, the mild reaction conditions reduce the energy load on manufacturing facilities and extend the lifespan of production equipment by minimizing corrosion and thermal stress. The simplified purification process reduces the consumption of solvents and consumables, directly contributing to cost reduction in pharmaceutical intermediates manufacturing without compromising on quality. For supply chain heads, this translates to a more predictable production schedule and reduced lead time for high-purity pharmaceutical intermediates, ensuring continuous availability for critical drug manufacturing pipelines.
- Cost Reduction in Manufacturing: The elimination of expensive and difficult-to-handle catalysts like sodium amide or specialized oxidants drastically simplifies the reagent procurement process and lowers raw material costs. By avoiding long-time reflux reactions and complex purification steps, the method reduces energy consumption and labor hours associated with batch processing. The use of common solvents and standard workup procedures further minimizes waste disposal costs, creating a leaner manufacturing model that enhances overall profit margins. This qualitative improvement in process efficiency allows for competitive pricing strategies while maintaining high quality standards essential for regulatory compliance.
- Enhanced Supply Chain Reliability: The use of readily available raw materials ensures that production is not held hostage by the scarcity of niche chemicals, thereby stabilizing the supply chain against market fluctuations. The robustness of the reaction conditions means that production can be maintained consistently across different facilities without requiring extensive re-validation or specialized training. This reliability is crucial for reducing lead time for high-purity pharmaceutical intermediates, allowing manufacturers to respond quickly to market demands and avoid stockouts. The simplified logistics of sourcing and storing reagents also reduce the risk of supply disruptions, ensuring a steady flow of materials for continuous commercial production.
- Scalability and Environmental Compliance: The method's low equipment requirements and mild conditions make it highly scalable from pilot plants to full commercial production without significant capital investment. The reduction in hazardous waste generation and the use of environmentally friendlier reagents align with modern green chemistry principles, facilitating easier regulatory approval and environmental compliance. This scalability ensures that the commercial scale-up of complex pharmaceutical intermediates can be achieved smoothly, meeting the growing global demand for lenalidomide. The process design inherently supports sustainable manufacturing practices, which is increasingly becoming a key criterion for supplier selection by major pharmaceutical companies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation and benefits of this synthesis technology. These answers are derived directly from the technical specifications and beneficial effects outlined in the patent documentation, providing clarity on process feasibility and quality control. Understanding these aspects is vital for stakeholders evaluating the integration of this route into their existing manufacturing portfolios. The responses highlight the practical advantages in terms of yield, safety, and operational simplicity that distinguish this method from conventional alternatives.
Q: What are the primary advantages of this synthesis route over prior art?
A: This method utilizes readily available raw materials like 3-nitrophthalic acid and avoids harsh conditions such as long-time reflux or difficult-to-prepare catalysts like sodium amide, resulting in easier purification and better industrial feasibility.
Q: How is the purity of the intermediate controlled during oxidation?
A: The process employs a selective oxidation step using copper salts and tert-butyl hydroperoxide at controlled temperatures between 50°C and 90°C, which minimizes excessive oxidation by-products and ensures high structural integrity.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the reaction conditions are mild, equipment requirements are low, and the post-treatment involves standard extraction and pulping techniques, making it highly adaptable for commercial scale-up of complex pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-(4-nitro-1-oxo-1,3-dihydro-2H-isoindol-2-yl)piperidine-2,6-dione Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust synthetic routes in the development of life-saving oncology therapies. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative methods like the one described in CN111548341B can be seamlessly transitioned from the lab to the plant. We are committed to maintaining stringent purity specifications and operating rigorous QC labs to guarantee that every batch of intermediate meets the highest industry standards. Our infrastructure is designed to handle complex chemistries with precision, providing a secure foundation for your drug development pipeline and ensuring consistent quality for your final API.
We invite you to collaborate with us to optimize your supply chain and leverage the commercial advantages of this advanced synthesis method. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific production needs, demonstrating how this route can enhance your operational efficiency. We encourage you to contact us to request specific COA data and route feasibility assessments, allowing you to make informed decisions based on concrete technical evidence. Partnering with us ensures access to a reliable pharmaceutical intermediates supplier dedicated to supporting your long-term growth and success in the global market.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →