Advanced Synthesis of Prucalopride Degradation Impurities for Global Quality Control
The pharmaceutical industry relies heavily on the stability and purity of active pharmaceutical ingredients (APIs) to ensure patient safety and regulatory compliance. Prucalopride, a selective serotonin 4 (5-HT4) receptor agonist used for treating chronic constipation, is known to exhibit sensitivity to light and oxidative conditions, leading to the formation of specific degradation products. Patent CN103755689A addresses a critical gap in quality control by disclosing a robust preparation method for a key degradation impurity, specifically 4-amino-N-[1-(3-methoxypropyl)-4-piperidinyl]-2,3-dihydrobenzofuran-7-carboxamide. This technical breakthrough allows manufacturers to move away from unreliable isolation methods towards a deterministic synthetic approach. By establishing a reliable supply of this specific impurity standard, pharmaceutical companies can significantly enhance their stability testing protocols and ensure that their final drug products meet the stringent purity specifications required by global health authorities. The ability to synthesize this compound on demand rather than relying on natural degradation represents a paradigm shift in how we approach impurity management for complex benzofuran derivatives.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, obtaining degradation impurities for reference standards involved subjecting the API to forced degradation conditions, such as intense light exposure or harsh oxidative environments, followed by complex chromatographic separation. As illustrated in the degradation pathway below, Prucalopride is extremely sensitive to light, which leads to the formation of the dechlorinated product. However, relying on this natural degradation process is fraught with inefficiencies and inconsistencies. The resulting mixtures are often complex, containing multiple unknown by-products that make the isolation of the target impurity difficult and costly. Furthermore, the yield from forced degradation is typically low and unpredictable, making it impossible to scale up for commercial quality control needs. This dependency on unstable degradation pathways creates a bottleneck in the supply chain for reference standards, potentially delaying regulatory filings and batch release testing. The lack of a controlled synthetic route means that laboratories are often at the mercy of variable degradation rates, leading to potential shortages of critical quality control materials.
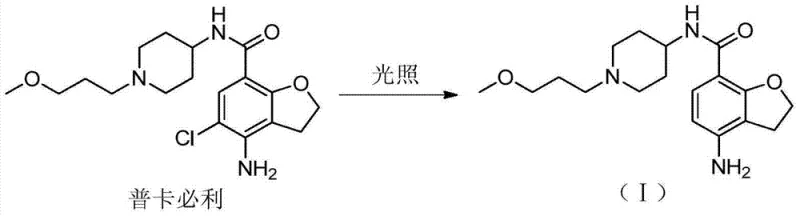
The Novel Approach
The novel approach detailed in the patent data circumvents these issues by employing a direct catalytic hydrogenation strategy to synthesize the impurity from the parent API. Instead of waiting for the molecule to break down unpredictably, this method intentionally and selectively removes the chlorine atom under controlled conditions. This shift from passive isolation to active synthesis provides a level of precision that is essential for modern pharmaceutical manufacturing. By using Prucalopride as the starting material, the process leverages an already available and high-purity feedstock, simplifying the raw material sourcing strategy. The reaction conditions are mild and utilize standard equipment found in most fine chemical facilities, ensuring that the method is not just theoretically sound but practically viable for commercial production. This approach guarantees a consistent supply of the impurity standard, independent of storage conditions or batch variability, thereby securing the quality control pipeline for manufacturers producing Prucalopride formulations globally.
Mechanistic Insights into Pd-C Catalyzed Hydrodechlorination
The core of this synthetic innovation lies in the mechanism of catalytic hydrodechlorination using a palladium-based catalyst. The reaction proceeds through the adsorption of the chlorinated benzofuran ring onto the surface of the palladium carbon catalyst. In the presence of hydrogen gas, the carbon-chlorine bond is cleaved, and a hydrogen atom is inserted in its place. This transformation is highly selective, leaving the sensitive amide bond and the piperidine ring intact, which is crucial for maintaining the structural integrity of the impurity relative to the parent drug. The use of a heterogeneous catalyst like 10% Pd-C allows for easy removal of the catalyst post-reaction via simple filtration, minimizing metal contamination in the final product. This mechanistic pathway is superior to chemical reduction methods that might require harsh reagents capable of damaging other functional groups. The specificity of the Pd-C catalyst ensures that the resulting compound is chemically identical to the photodegradation product, validating its use as a reference standard for stability studies.
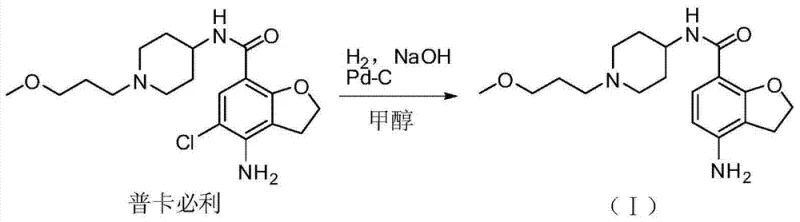
Impurity control is further enhanced by the addition of an acid-binding agent, such as sodium hydroxide, during the reaction. The hydrodechlorination process generates hydrochloric acid as a by-product, which, if left unchecked, could catalyze unwanted side reactions or lead to the formation of salts that complicate purification. By neutralizing this acid in situ, the reaction environment remains stable, promoting higher conversion rates and cleaner product profiles. The patent data indicates that operating at temperatures between 10-30°C optimizes this balance, preventing thermal degradation while maintaining sufficient reaction kinetics. This careful control of the reaction milieu ensures that the impurity profile remains simple, facilitating downstream purification. For R&D directors, this means that the synthetic route not only produces the target molecule but does so with a purity profile that simplifies analytical method validation and regulatory documentation.
How to Synthesize 4-Amino-N-[1-(3-methoxypropyl)-4-piperidinyl]-2,3-dihydrobenzofuran-7-carboxamide Efficiently
The synthesis of this critical degradation impurity is designed to be operationally simple while maintaining high standards of reproducibility. The process begins by dissolving the Prucalopride starting material in a lower alcohol solvent, with methanol being the preferred choice due to its excellent solubility properties and compatibility with the hydrogenation catalyst. The addition of the base and catalyst must be managed carefully to ensure uniform dispersion before introducing hydrogen gas. The detailed standardized synthesis steps see below guide.
- Dissolve Prucalopride starting material in a lower alcohol solvent such as methanol or isopropanol within a hydrogenation reactor.
- Add a heterogeneous metal catalyst, specifically 10% Palladium on Carbon (Pd-C), along with an inorganic base like sodium hydroxide to act as an acid scavenger.
- Conduct the hydrogenation reaction under atmospheric pressure at a controlled temperature range of 10-30°C for approximately 72 hours, followed by filtration and concentration.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this synthetic method offers substantial strategic advantages beyond mere technical feasibility. The ability to produce this impurity on demand eliminates the need for maintaining large inventories of unstable degradation mixtures or relying on external suppliers who may use inconsistent isolation methods. This shift significantly reduces the risk of supply disruptions that could halt quality control testing. Furthermore, the use of common reagents like methanol, hydrogen, and palladium carbon means that the cost of goods sold for this impurity is driven by commodity prices rather than specialized, scarce reagents. This stability in raw material sourcing translates directly into predictable budgeting and reduced exposure to market volatility for critical quality control consumables.
- Cost Reduction in Manufacturing: The elimination of complex chromatographic separation steps, which are typically required to isolate impurities from degradation mixtures, results in significant processing cost savings. Traditional isolation often requires preparative HPLC, which is solvent-intensive and time-consuming. By contrast, the hydrogenation method allows for simple filtration and concentration, drastically reducing solvent consumption and labor hours. Additionally, the high molar yield reported in the patent examples suggests that raw material utilization is optimized, minimizing waste. This efficiency gain allows manufacturers to allocate resources more effectively, focusing on core production activities rather than struggling with low-yield impurity isolation processes that drain operational budgets.
- Enhanced Supply Chain Reliability: Relying on a synthetic route ensures that the supply of reference standards is decoupled from the stability limitations of the API itself. Since the impurity is generated from fresh API stock, there is no risk of the starting material degrading before use. This reliability is crucial for maintaining continuous quality control operations, especially for long-term stability studies that require consistent standards over months or years. The robustness of the hydrogenation process means that production can be scaled up or down based on demand without compromising quality. This flexibility provides supply chain heads with the confidence to plan long-term quality assurance strategies without the fear of sudden material shortages.
- Scalability and Environmental Compliance: The process utilizes standard hydrogenation equipment that is readily available in most pharmaceutical manufacturing facilities, eliminating the need for capital investment in specialized isolation machinery. The reaction conditions are mild and operate at atmospheric pressure, reducing energy consumption and safety risks associated with high-pressure systems. Furthermore, the use of a heterogeneous catalyst allows for recovery and potential recycling of the palladium, aligning with green chemistry principles and reducing heavy metal waste. The simplified workup procedure minimizes the generation of hazardous waste streams, making it easier to comply with increasingly stringent environmental regulations. This scalability ensures that the method remains viable from gram-scale R&D to multi-kilogram commercial production.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of this Prucalopride degradation impurity. These answers are derived directly from the patent specifications and are intended to clarify the implementation of this technology in a commercial setting. Understanding these details is essential for technical teams evaluating the feasibility of adopting this synthetic route for their internal quality control laboratories or external supply partnerships.
Q: What is the primary chemical transformation in this impurity synthesis?
A: The core transformation is a catalytic hydrodechlorination where the chlorine atom on the benzofuran ring of Prucalopride is replaced by a hydrogen atom using Pd-C and hydrogen gas.
Q: Why is sodium hydroxide added during the hydrogenation process?
A: Sodium hydroxide acts as an acid-binding agent to neutralize the hydrochloric acid generated during the dechlorination, thereby preventing side reactions and improving the overall molar yield of the target impurity.
Q: How does this synthetic method improve supply chain stability for reference standards?
A: By synthesizing the impurity directly from the API rather than isolating it from unstable degradation mixtures, manufacturers can ensure a consistent, high-purity supply of reference standards independent of storage conditions.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Prucalopride Impurity Supplier
The synthesis route described in patent CN103755689A represents a significant advancement in the management of pharmaceutical impurities, offering a reliable and scalable solution for global quality control needs. NINGBO INNO PHARMCHEM stands ready to support your organization in implementing this technology, leveraging our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facility is equipped with state-of-the-art hydrogenation reactors and rigorous QC labs capable of meeting stringent purity specifications required for reference standards. We understand the critical nature of impurity management in the pharmaceutical lifecycle and are committed to delivering materials that ensure your regulatory compliance and product safety.
We invite you to engage with our technical procurement team to discuss how this synthetic method can optimize your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching from isolation to synthesis. We encourage potential partners to contact us for specific COA data and route feasibility assessments tailored to your production volumes. Let us help you secure a stable, high-quality supply of critical impurity standards, ensuring that your Prucalopride products maintain the highest levels of quality and reliability in the global market.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →