Advanced One-Step Synthesis of Amorphous Obeticholic Acid Form 1 for Commercial Scale-up
Advanced One-Step Synthesis of Amorphous Obeticholic Acid Form 1 for Commercial Scale-up
The pharmaceutical landscape for treating primary biliary cirrhosis and non-alcoholic fatty liver disease has been significantly advanced by the discovery of Farnesoid X Receptor (FXR) agonists, specifically Obeticholic Acid, yet the commercial viability of this potent therapeutic agent relies heavily on the robustness of its manufacturing process as detailed in patent CN105566429B. This intellectual property discloses a highly efficient preparation method for Obeticholic Acid Form 1, an amorphous solid form that has emerged as the preferred polymorph for active pharmaceutical ingredient (API) development due to its favorable physicochemical properties compared to various crystalline counterparts. The technical breakthrough lies in the ability to bypass complex multi-step crystallization sequences, directly yielding the target amorphous material through a streamlined reduction protocol that utilizes readily available industrial reagents. For R&D directors and procurement specialists evaluating reliable pharmaceutical intermediates supplier partnerships, understanding the nuances of this direct synthesis route is critical for assessing long-term supply security and cost structures. The methodology not only addresses the historical challenges associated with polymorphic control but also introduces a scalable pathway that aligns with modern green chemistry principles by minimizing solvent usage and operational complexity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methods, such as those disclosed in patent CN104781272A, typically relied on a convoluted strategy that necessitated the preliminary preparation of high-purity crystalline Obeticholic Acid Form C before any conversion to the desired amorphous Form 1 could occur. This traditional approach introduced significant bottlenecks in the manufacturing workflow, including the requirement for high-boiling point solvents that are difficult to recover and recycle, leading to inflated operational expenditures and environmental burdens. Furthermore, the necessity to isolate and purify an intermediate crystalline form added multiple unit operations to the production line, such as additional filtration, drying, and re-dissolution steps, which inherently increased the risk of yield loss and cross-contamination. From a supply chain perspective, the reliance on specific crystalline intermediates created fragility; any deviation in the crystallization of Form C could halt the entire downstream process, resulting in prolonged production periods and inconsistent batch-to-batch quality. These inefficiencies rendered the conventional routes economically unviable for large-scale commercial production, prompting the industry to seek a more direct and robust synthetic alternative that could deliver high-purity material without the baggage of intermediate isolation.
The Novel Approach
The innovative methodology presented in CN105566429B fundamentally disrupts this status quo by enabling the direct transformation of the 7-keto precursor into Obeticholic Acid Form 1 in a single, cohesive operation. By leveraging a specific alkaline reduction environment, the process achieves the simultaneous reduction of the ketone functionality and the formation of the amorphous solid state upon workup, effectively collapsing what was once a multi-stage sequence into a simplified workflow. This novel approach eliminates the prerequisite for generating crystalline Form C, thereby removing the associated costs of specialized solvents and extended processing times. The result is a manufacturing protocol that is not only chemically elegant but also commercially superior, offering a drastic simplification of the operational footprint. For procurement managers focused on cost reduction in pharmaceutical intermediates manufacturing, this shift represents a tangible opportunity to lower the cost of goods sold (COGS) by reducing reagent consumption, energy usage, and labor hours associated with complex purification steps. The ability to obtain the final API form directly from the reduction reaction mixture underscores the process's maturity and readiness for industrial adoption.
Mechanistic Insights into Alkaline Hydride Reduction
The core chemical transformation driving this synthesis is the stereoselective reduction of the 7-ketone group on the cholane steroid backbone to the corresponding 7-alpha-hydroxyl group, a reaction that demands precise control over reaction conditions to ensure the correct stereochemistry and polymorphic outcome. The process utilizes alkali metal borohydrides, such as sodium borohydride or potassium borohydride, in a heated aqueous or alcoholic alkaline medium, typically maintained between 75°C and 105°C to facilitate rapid kinetics and complete conversion of the starting material. The presence of a strong base, such as sodium hydroxide or sodium alkoxide, is crucial not only for solubilizing the acidic substrate but also for modulating the reactivity of the hydride species, ensuring that the reduction proceeds cleanly without affecting other sensitive functional groups on the molecule. Following the reduction phase, the reaction mixture is carefully quenched and subjected to a pH-controlled extraction, where the pH is adjusted to a mildly acidic range of 3 to 5 to protonate the carboxylic acid moiety, allowing for efficient partitioning into an organic phase. This meticulous control over pH and temperature during the workup phase is instrumental in preventing the formation of unwanted crystalline polymorphs, steering the molecule towards the thermodynamically less stable but pharmaceutically desirable amorphous state.
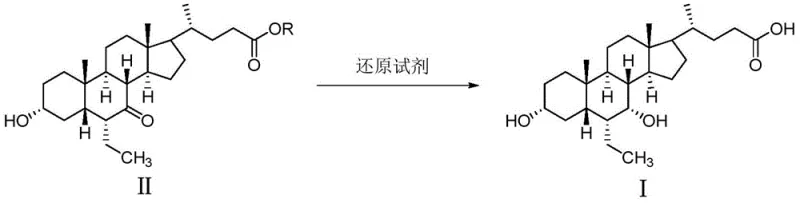
Impurity control within this mechanistic framework is achieved through the strategic selection of solvents and the implementation of a recrystallization-like precipitation step that acts as a powerful purification driver. The patent specifies the use of common organic solvents like ethyl acetate, dichloromethane, or toluene for extraction, which are effective at separating the product from inorganic salts and polar byproducts generated during the reduction. Subsequent dissolution of the oily residue in water with alkali, followed by slow dropwise addition of dilute hydrochloric acid, creates a supersaturated environment that favors the nucleation of the amorphous Form 1 while excluding impurities that might otherwise co-crystallize. This "dissolve-and-precipitate" technique serves as a final polishing step, ensuring that the resulting solid meets stringent purity specifications, often exceeding 99.5%, which is critical for meeting regulatory standards for high-purity pharmaceutical intermediates. The robustness of this mechanism allows for significant flexibility in scaling, as the fundamental chemical drivers remain consistent regardless of batch size, provided that mixing and heat transfer are adequately managed.
How to Synthesize Obeticholic Acid Form 1 Efficiently
The practical execution of this synthesis involves a sequence of well-defined unit operations that can be readily implemented in standard multipurpose chemical reactors, making it accessible for contract development and manufacturing organizations (CDMOs) aiming to support clinical and commercial supply. The process begins with the charging of the 7-keto precursor and base into a reactor, followed by heating and the controlled addition of the reducing agent, a step that requires careful monitoring via TLC to confirm reaction completion before proceeding to workup. Once the reduction is verified, the mixture undergoes a phase separation and solvent swap, culminating in a controlled acidification and cooling cycle that triggers the precipitation of the target amorphous solid. For technical teams looking to implement this route, the detailed standardized synthesis steps are provided below to ensure reproducibility and adherence to the patented parameters.
- Heat a mixture of 3-alpha-hydroxy-6-alpha-ethyl-7-keto-5-beta-cholane-24-acid (or ester), alkali, and solvent to 75-105°C, then add a reducing agent like sodium borohydride to complete the reduction.
- Cool the reaction mixture, extract with organic solvent, adjust pH to 3-5, separate layers, and evaporate to obtain an oily residue which is then dissolved in water and alkali.
- Slowly add dilute hydrochloric acid to the solution to adjust pH to 3-4, cool to 10-15°C to induce crystallization, filter the solid, and vacuum dry to obtain the final product.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, the adoption of the CN105566429B process offers profound advantages that extend beyond simple chemical yield, impacting the overall resilience and economics of the supply chain for Obeticholic Acid. By eliminating the need for intermediate isolation and the use of exotic high-boiling solvents, the process inherently reduces the complexity of the manufacturing infrastructure required, allowing for faster turnaround times and greater agility in responding to market demand fluctuations. The reliance on commodity chemicals like sodium borohydride and hydrochloric acid, rather than specialized catalysts or reagents, mitigates the risk of supply disruptions and price volatility, ensuring a more stable and predictable cost base for long-term procurement planning. Furthermore, the simplified workflow reduces the total number of processing days per batch, effectively increasing the throughput capacity of existing manufacturing facilities without the need for significant capital investment in new equipment. These factors combine to create a supply model that is not only cost-effective but also highly reliable, addressing the critical needs of supply chain heads who prioritize continuity and risk mitigation in their vendor selection criteria.
- Cost Reduction in Manufacturing: The elimination of the intermediate crystalline Form C isolation step results in substantial cost savings by removing entire unit operations such as filtration, drying, and re-dissolution, which are both labor-intensive and energy-consuming. Additionally, the use of low-cost, easily recoverable solvents like ethyl acetate and water significantly lowers the raw material expenditure compared to processes requiring expensive chlorinated or high-boiling solvents that are difficult to recycle. The overall reduction in process time and resource consumption translates directly into a lower cost of goods, providing a competitive pricing advantage for buyers seeking commercial scale-up of complex pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: The robustness of the direct reduction method minimizes the potential for batch failures associated with complex crystallization steps, thereby enhancing the reliability of supply and reducing the lead time for delivering high-purity materials. Since the process does not depend on the availability of specific crystalline seeds or hard-to-source reagents, manufacturers can maintain consistent production schedules even in the face of minor raw material variations. This operational stability is crucial for pharmaceutical companies that require guaranteed supply continuity to support clinical trials and commercial launches without interruption.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing reaction conditions and workup procedures that are easily transferable from pilot plant to full commercial scale, facilitating the commercial scale-up of complex APIs. Moreover, the reduced solvent load and the ability to recycle aqueous and organic phases align with increasingly stringent environmental regulations, lowering the cost and complexity of waste treatment and disposal. This environmental efficiency not only reduces operational overhead but also enhances the sustainability profile of the supply chain, a key consideration for modern pharmaceutical procurement strategies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of Obeticholic Acid Form 1, derived directly from the specific embodiments and comparative data found within the patent literature. These insights are intended to clarify the operational benefits and technical feasibility of the described method for stakeholders evaluating potential manufacturing partners. Understanding these details is essential for making informed decisions about technology transfer and long-term supply agreements.
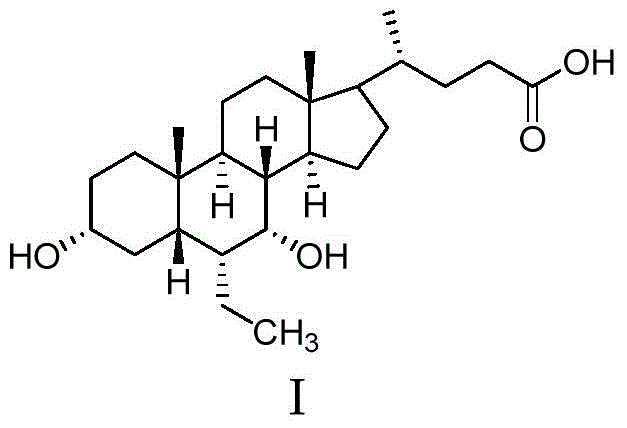
Q: Why is Obeticholic Acid Form 1 preferred over crystalline forms?
A: Obeticholic Acid Form 1 is an amorphous solid form that offers superior processing characteristics compared to crystalline forms like C, D, F, and G, which have been found unsuitable for further pharmaceutical development due to stability and bioavailability issues.
Q: What is the primary advantage of the method in CN105566429B?
A: The primary advantage is the direct one-step conversion of the 7-keto precursor to the amorphous Form 1, eliminating the need to first synthesize and isolate high-purity crystalline Form C, thereby drastically simplifying the workflow.
Q: Which reducing agents are compatible with this synthesis?
A: The process supports the use of common alkali metal borohydrides, specifically sodium borohydride, potassium borohydride, or sodium cyanoborohydride, allowing for flexibility in reagent sourcing and cost management.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Obeticholic Acid Form 1 Supplier
As a premier CDMO partner, NINGBO INNO PHARMCHEM possesses the technical expertise and infrastructure necessary to translate the innovative synthesis route described in CN105566429B into a reality for global pharmaceutical clients. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and compliant with all regulatory standards. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of Obeticholic Acid Form 1 meets the highest quality benchmarks required for API manufacturing. Our commitment to excellence ensures that clients receive a product that is not only chemically pure but also consistent in its physical properties, critical for the efficacy and safety of the final drug product.
We invite potential partners to engage with our technical procurement team to discuss how this advanced manufacturing process can be tailored to meet your specific volume and timeline requirements. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits of switching to this streamlined synthesis route. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing you to move forward with confidence in securing a reliable and cost-effective supply of this critical therapeutic agent.