Advanced Synthesis of Bioactive Urea Derivatives for Oncology Applications
The pharmaceutical landscape is constantly evolving with the demand for novel scaffolds that exhibit potent biological activity while maintaining synthetic feasibility. Patent CN101161645B introduces a significant advancement in the field of medicinal chemistry by disclosing a series of novel urea derivatives characterized by a specific general formula. These compounds are not merely theoretical constructs but have been rigorously tested and shown to possess obvious inhibitory effects on critical cancer cell lines, specifically the human oral epidermal cancer cell line (KB) and the human leukemia cell line (K562). The core innovation lies in the structural versatility allowed by the R1 and R2 substituents, which can be tailored to optimize pharmacokinetic properties and binding affinity. This patent provides a robust foundation for developing next-generation antitumor agents, offering a clear pathway from molecular design to therapeutic application.
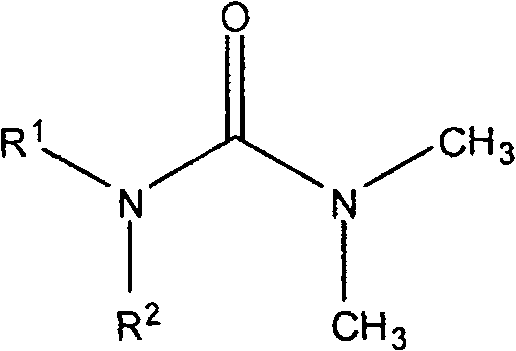
For procurement managers and supply chain heads, the significance of this technology extends beyond biological efficacy to manufacturability. The ability to systematically vary the R1 group, which includes substituted benzyl moieties such as nitro, chloro, and hydroxy variants, allows for a modular approach to library synthesis. This modularity is crucial for a reliable pharmaceutical intermediate supplier, as it enables the rapid generation of analogues to address specific structure-activity relationship (SAR) requirements without necessitating entirely new process development for each candidate. The patent explicitly details the preparation methods, ensuring that the intellectual property is supported by reproducible experimental data, which is a key indicator of technological maturity for potential licensing or commercial partnership.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the synthesis of N-substituted urea derivatives has been fraught with challenges that hinder efficient commercial scale-up. Conventional methods often rely on the use of isocyanates, which are highly toxic, moisture-sensitive, and hazardous to handle on a large industrial scale. The requirement for strict anhydrous conditions and specialized containment equipment significantly drives up capital expenditure and operational costs. Furthermore, alternative coupling strategies using carbonyl diimidazole (CDI) or similar activating agents, while effective on a milligram scale, become prohibitively expensive when transitioning to kilogram or ton-scale production. These reagents generate stoichiometric amounts of waste byproducts that require complex disposal protocols, adding to the environmental burden and reducing the overall atom economy of the process. Additionally, traditional thermal methods often require elevated temperatures that can lead to the decomposition of sensitive functional groups, limiting the scope of substrates that can be successfully utilized.
The Novel Approach
The methodology disclosed in patent CN101161645B offers a transformative solution to these longstanding issues by employing an in-situ activation strategy using dimethylformamide (DMF) and thionyl chloride (SOCl2). This approach generates a highly reactive chloro-dimethyl-formamidinium intermediate under controlled conditions, effectively mimicking the reactivity of more dangerous reagents without the associated risks. The process operates at moderate temperatures, typically between 50-60°C for the coupling step, which preserves the integrity of sensitive substituents like nitro and hydroxy groups found on the aromatic rings. By utilizing commodity chemicals that are readily available in the global chemical market, this novel route drastically simplifies the supply chain logistics. The reaction proceeds with high efficiency, yielding target compounds such as N,N-dimethyl-N'-(3-morpholinopropyl)-N'-(3-nitrobenzyl)urea in respectable yields, demonstrating that high purity can be achieved without compromising on safety or cost-effectiveness.
Mechanistic Insights into DMF-Mediated Urea Formation
The core mechanistic advantage of this synthesis lies in the Vilsmeier-Haack type activation of the amide solvent. In the initial step, anhydrous DMF reacts with thionyl chloride in a molar ratio ranging from 1:2 to 1:2.5 within an anhydrous dichloromethane medium. Upon heating to 70-80°C, the oxygen of the DMF carbonyl attacks the sulfur of the thionyl chloride, leading to the elimination of SO2 and HCl and the formation of the electrophilic chloro-dimethyl-formamidinium chloride species. This intermediate is exceptionally reactive towards nucleophiles. In the subsequent step, a solution containing the target amine (R1-NH-R2) and pyridine is introduced dropwise. Pyridine serves a dual purpose: it acts as a base to neutralize the generated acid and as a nucleophilic catalyst to facilitate the attack of the amine nitrogen on the activated carbonyl carbon of the intermediate. This nucleophilic acyl substitution results in the formation of the urea linkage, releasing dimethylamine as a leaving group which is scavenged by the acidic byproducts.
Impurity control is meticulously managed through precise temperature regulation and workup procedures. The patent specifies heating the reaction mixture to 50-60°C for 5-6 hours, a window that ensures complete conversion while minimizing side reactions such as over-chlorination or degradation of the morpholine or piperidine rings present in the R2 substituents. Following the reaction, the mixture is quenched in ice water, a critical step that hydrolyzes any remaining unreacted activated intermediate, preventing it from reacting during the isolation phase. The organic phase is separated and washed with saturated sodium bicarbonate to remove acidic impurities, followed by drying over anhydrous sodium sulfate. The final purification via column chromatography using petroleum ether and ethyl acetate gradients ensures that the final product meets stringent purity specifications required for pharmaceutical applications, effectively removing trace amounts of starting amines or dialkylated byproducts.
How to Synthesize N-Substituted Urea Derivatives Efficiently
The synthesis of these high-value urea derivatives follows a standardized four-step protocol that balances reaction kinetics with operational safety. The process begins with the activation of the solvent system, followed by the controlled addition of the amine substrate, reaction maintenance, and finally, isolation and purification. This sequence has been optimized to maximize yield while minimizing the formation of difficult-to-remove impurities. For research and development teams looking to replicate or scale this chemistry, adherence to the specific molar ratios and temperature profiles outlined in the patent is essential for success. The detailed标准化 synthesis steps are provided in the guide below to ensure reproducibility across different laboratory settings.
- Activate anhydrous DMF with thionyl chloride (SOCl2) in dichloromethane at 70-80°C to form the reactive chloro-formamidinium intermediate.
- Prepare a solution of the target amine substrate (R1-NH-R2) and pyridine in anhydrous dichloromethane.
- Combine the activated intermediate with the amine solution, reacting at 50-60°C for 5-6 hours to form the urea bond.
- Perform aqueous workup followed by column chromatography to isolate the high-purity urea derivative.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthetic route offers substantial strategic benefits for organizations focused on cost reduction in pharmaceutical intermediate manufacturing. The primary driver of value is the replacement of expensive, specialized coupling reagents with bulk commodity chemicals. DMF and thionyl chloride are produced in massive volumes globally, ensuring a stable supply chain and shielding manufacturers from the price volatility often seen with niche fine chemical reagents. This shift fundamentally alters the cost structure of the synthesis, allowing for significant margin improvement or more competitive pricing for the final active pharmaceutical ingredient (API). Furthermore, the avoidance of toxic isocyanates reduces the regulatory burden and insurance costs associated with handling hazardous materials, contributing to a safer and more sustainable manufacturing environment.
- Cost Reduction in Manufacturing: The elimination of high-cost activating agents like CDI or HATB directly lowers the bill of materials. Since the process utilizes catalytic amounts of pyridine and stoichiometric amounts of inexpensive thionyl chloride, the overall reagent cost per kilogram of product is drastically reduced. Additionally, the simplified workup procedure, which relies on standard liquid-liquid extraction rather than complex crystallization or distillation setups, reduces energy consumption and labor hours. This efficiency translates into a leaner production process where resources are allocated more effectively, enhancing the overall economic viability of producing these complex urea scaffolds for oncology applications.
- Enhanced Supply Chain Reliability: Sourcing raw materials is a critical bottleneck in many synthetic routes, but this method mitigates that risk by relying on widely available solvents and reagents. Dichloromethane, DMF, and thionyl chloride are staple chemicals in the fine chemical industry, meaning multiple qualified suppliers exist in every major chemical hub. This redundancy ensures that production schedules are not disrupted by single-source failures or logistical delays. For supply chain heads, this reliability is paramount for maintaining continuous manufacturing operations and meeting delivery commitments to downstream pharmaceutical clients who depend on timely access to high-quality intermediates for their drug development pipelines.
- Scalability and Environmental Compliance: The process is inherently scalable due to its homogeneous nature and moderate thermal requirements. Moving from bench scale to pilot plant or commercial production does not require exotic reactor materials or extreme pressure conditions. The waste stream is also more manageable; while thionyl chloride generates acidic off-gases, these can be easily scrubbed using standard caustic scrubbers already present in compliant facilities. The absence of heavy metal catalysts further simplifies waste treatment and disposal, aligning with increasingly strict environmental regulations. This ease of scale-up ensures that the transition from clinical trial material to commercial supply can be executed smoothly without the need for extensive process re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of these urea derivatives. These answers are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and relevance for stakeholders evaluating this technology for potential integration into their own R&D or manufacturing portfolios. Understanding these nuances is critical for making informed decisions about process adoption.
Q: What is the biological activity profile of these urea derivatives?
A: The synthesized urea derivatives demonstrate significant inhibitory effects against human oral epidermal cancer cell lines (KB) and human leukemia cell lines (K562), making them valuable candidates for antitumor drug development.
Q: How does this synthesis method improve upon traditional urea formation?
A: This method utilizes an in-situ activation of DMF with thionyl chloride, avoiding the handling of hazardous isocyanates and enabling milder reaction conditions (50-60°C) compared to traditional high-temperature processes.
Q: Can this process be scaled for commercial manufacturing?
A: Yes, the process relies on commodity chemicals like DMF and SOCl2 and utilizes standard extraction and chromatography techniques, facilitating straightforward scale-up from laboratory to industrial production volumes.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Urea Derivatives Supplier
As the demand for specialized oncology intermediates grows, partnering with an experienced CDMO becomes a strategic imperative. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can seamlessly transition from discovery to market. Our facility is equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of urea derivatives meets the highest international standards. We understand the critical nature of time-to-market in the pharmaceutical sector and are committed to providing a seamless, compliant, and efficient manufacturing partnership.
We invite you to engage with our technical procurement team to discuss how we can support your specific needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into how optimizing this synthetic route can benefit your bottom line. We encourage potential partners to contact us for specific COA data and route feasibility assessments tailored to your target molecules. Let us leverage our expertise in complex organic synthesis to accelerate your drug development timeline and secure your supply chain for the future.