Advanced Manufacturing of Carbocyclic Purine Antiviral Intermediates via Salt-Mediated Alkylation
The pharmaceutical industry continuously seeks robust and scalable synthetic routes for potent antiviral agents, particularly those targeting herpes viruses and HIV. Patent CN1036712C presents a significant technological advancement in the preparation of carbocyclic purine derivatives, specifically focusing on the synthesis of [1R-(1α,2β,3α)]-2-amino-9-[2,3-bis(hydroxymethyl)cyclobutyl]-1,9-dihydro-6H-purin-6-one. This compound serves as a critical intermediate or active pharmaceutical ingredient with broad-spectrum antiviral activity. The core innovation lies in the utilization of specific purine salts to facilitate nucleophilic substitution on activated cyclobutane rings, overcoming the solubility and reactivity limitations often encountered with free purine bases. ![Chemical structure of the target antiviral agent [1R-(1α,2β,3α)]-2-amino-9-[2,3-bis(hydroxymethyl)cyclobutyl]-1,9-dihydro-6H-purin-6-one](/insights/img/antiviral-purine-intermediate-synthesis-supplier-20260305211459-01.png) . By shifting from traditional harsh alkylation conditions to a salt-mediated approach, the process achieves superior stereocontrol and yield, making it highly attractive for commercial scale-up in the production of high-purity pharmaceutical intermediates.
. By shifting from traditional harsh alkylation conditions to a salt-mediated approach, the process achieves superior stereocontrol and yield, making it highly attractive for commercial scale-up in the production of high-purity pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the alkylation of purine bases with carbohydrate surrogates has been plagued by several significant technical hurdles that impact both cost and quality. Traditional methods often rely on the use of strong, inorganic bases such as sodium hydride or potassium carbonate in polar aprotic solvents like DMF. These conditions frequently lead to poor regioselectivity, resulting in mixtures of N7 and N9 alkylated isomers that are difficult and expensive to separate. Furthermore, the harsh basic environment can cause degradation of sensitive protecting groups on the sugar surrogate or induce elimination reactions on the cyclobutane ring, leading to olefinic impurities. The low solubility of free purine bases in common organic solvents also necessitates large solvent volumes and extended reaction times, which reduces throughput and increases waste generation. These inefficiencies create substantial bottlenecks for procurement managers seeking reliable suppliers capable of delivering consistent quality at a competitive price point.
The Novel Approach
The methodology disclosed in the patent introduces a paradigm shift by employing pre-formed purine salts, such as the tetrabutylammonium or benzyltriethylammonium salts of 6-halo-9H-purin-2-amine. This strategy dramatically enhances the nucleophilicity of the purine nitrogen while maintaining excellent solubility in moderately polar organic solvents like dichloromethane or acetonitrile. 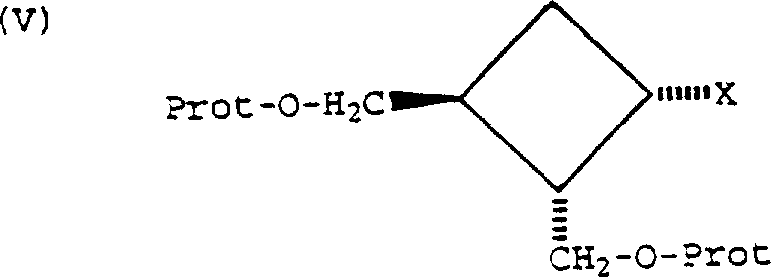 . The reaction proceeds under remarkably mild conditions, often between 0°C and 30°C when using triflate leaving groups, which minimizes thermal degradation and side reactions. This approach not only simplifies the workup procedure by avoiding the neutralization of strong inorganic bases but also ensures high regioselectivity for the desired N9-alkylated product. For supply chain heads, this translates to a more predictable manufacturing timeline and reduced dependency on specialized equipment capable of handling hazardous reagents.
. The reaction proceeds under remarkably mild conditions, often between 0°C and 30°C when using triflate leaving groups, which minimizes thermal degradation and side reactions. This approach not only simplifies the workup procedure by avoiding the neutralization of strong inorganic bases but also ensures high regioselectivity for the desired N9-alkylated product. For supply chain heads, this translates to a more predictable manufacturing timeline and reduced dependency on specialized equipment capable of handling hazardous reagents.
Mechanistic Insights into Salt-Mediated Nucleophilic Substitution
The core chemical transformation involves a bimolecular nucleophilic substitution (SN2) mechanism where the N9 nitrogen of the purine salt attacks the electrophilic carbon of the cyclobutane ring bearing the leaving group. The use of a quaternary ammonium cation serves a dual purpose: it acts as a phase-transfer catalyst equivalent by solubilizing the purine anion in the organic phase and stabilizes the transition state through ion-pairing effects. 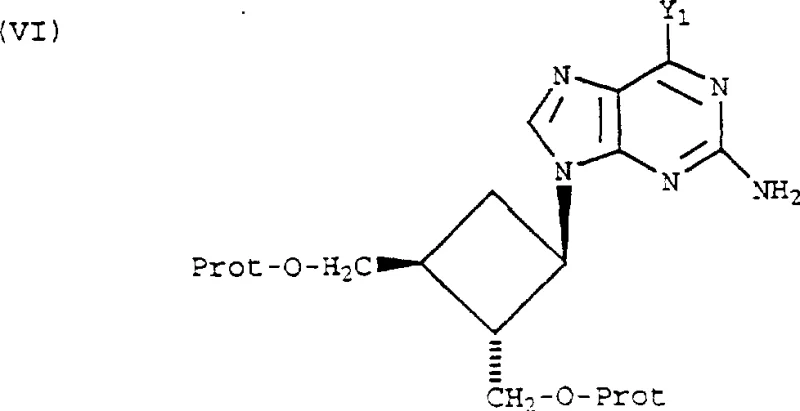 . The stereochemical outcome is strictly governed by the configuration of the starting cyclobutane derivative; since the leaving group X is cis to the adjacent hydroxymethyl group, the backside attack by the purine results in an inversion of configuration, yielding the trans-disposed relationship required for biological activity. This precise control is critical for R&D directors who must ensure that the impurity profile remains within stringent regulatory limits. The choice of leaving group, whether it be a trifluoromethanesulfonate or a p-nitrobenzenesulfonate, allows for fine-tuning the reaction kinetics to match the specific stability profile of the protecting groups employed.
. The stereochemical outcome is strictly governed by the configuration of the starting cyclobutane derivative; since the leaving group X is cis to the adjacent hydroxymethyl group, the backside attack by the purine results in an inversion of configuration, yielding the trans-disposed relationship required for biological activity. This precise control is critical for R&D directors who must ensure that the impurity profile remains within stringent regulatory limits. The choice of leaving group, whether it be a trifluoromethanesulfonate or a p-nitrobenzenesulfonate, allows for fine-tuning the reaction kinetics to match the specific stability profile of the protecting groups employed.
Following the coupling step, the conversion to the final antiviral agent involves a carefully orchestrated sequence of deprotection and hydrolysis. The patent details multiple pathways to remove the hydroxyl protecting groups (such as benzoyl or silyl groups) and convert the 6-halo or 6-methoxy substituent into the requisite 6-oxo function. For instance, treatment with sodium methoxide in refluxing methanol effectively displaces the 6-iodo group with a methoxy group while simultaneously cleaving ester protecting groups. Subsequent acid hydrolysis then converts the 6-methoxy intermediate into the final 6-oxo purine. This modularity allows manufacturers to select the most cost-effective protecting group strategy without compromising the final step's efficiency. Understanding these mechanistic nuances is essential for optimizing the process to reduce solvent consumption and energy usage, directly impacting the overall cost of goods sold.
How to Synthesize Carbocyclic Purine Antiviral Agents Efficiently
The synthesis of these complex molecules requires precise control over reaction parameters to ensure high purity and yield. The patented process outlines a clear pathway starting from readily available purine halides and protected cyclobutanols. The initial formation of the purine salt is a critical unit operation that sets the stage for the subsequent coupling efficiency. Detailed standard operating procedures for each step, including specific molar ratios, temperature profiles, and crystallization protocols, are essential for technology transfer. . Implementing these standardized steps ensures reproducibility across different manufacturing sites and scales, which is a key requirement for qualifying as a reliable supplier in the global pharmaceutical market.
- Preparation of the purine salt by reacting 6-halo-9H-purin-2-amine with a quaternary ammonium hydroxide in a suitable solvent system.
- Coupling the purine salt with a protected bis(hydroxymethyl)cyclobutane derivative containing a good leaving group (e.g., triflate or nosylate) under mild conditions.
- Selective deprotection of hydroxyl groups and hydrolysis of the 6-substituent to yield the final 6-oxo antiviral agent.
Commercial Advantages for Procurement and Supply Chain Teams
Adopting this salt-mediated alkylation technology offers profound commercial benefits that extend beyond simple chemical yield improvements. For procurement managers, the ability to use milder reaction conditions means a significant reduction in energy costs associated with heating and cooling large reactors. The avoidance of hazardous strong bases like sodium hydride also lowers safety compliance costs and insurance premiums, contributing to substantial cost savings in manufacturing. Furthermore, the high regioselectivity of the reaction minimizes the formation of difficult-to-remove isomers, reducing the need for extensive chromatographic purification which is often a major cost driver in API production. This efficiency allows for a more streamlined supply chain with fewer processing steps and shorter cycle times.
- Cost Reduction in Manufacturing: The elimination of expensive and hazardous reagents, combined with the use of common solvents like dichloromethane and acetonitrile, drastically simplifies the raw material procurement process. The high conversion rates observed in the examples suggest that less starting material is wasted, leading to better atom economy. Additionally, the simplified workup procedures, which often involve straightforward filtration or extraction rather than complex distillations, reduce labor and utility costs. These factors collectively drive down the production cost, making the final antiviral intermediate more competitive in the marketplace.
- Enhanced Supply Chain Reliability: The robustness of the chemical process against minor variations in temperature and stoichiometry ensures consistent batch-to-batch quality. This reliability is crucial for maintaining continuous supply to downstream API manufacturers. The use of stable intermediates, such as the protected cyclobutane triflates which can be stored or generated in situ, provides flexibility in production scheduling. By reducing the risk of batch failures due to side reactions, manufacturers can guarantee shorter lead times for high-purity pharmaceutical intermediates, thereby strengthening the overall resilience of the supply chain.
- Scalability and Environmental Compliance: The process is inherently scalable, as demonstrated by the successful execution of reactions on multi-gram scales in the patent examples without loss of efficiency. The reduced generation of inorganic salt waste, typical of traditional base-mediated alkylations, aligns with modern green chemistry principles and environmental regulations. This makes the technology easier to permit and operate in jurisdictions with strict waste disposal laws. The ability to scale from pilot plant to commercial production with minimal process re-engineering ensures that supply can meet growing market demand for antiviral therapies without significant capital investment.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These insights are derived directly from the experimental data and claims within the patent documentation. . Addressing these points proactively helps potential partners understand the feasibility and advantages of integrating this technology into their existing manufacturing portfolios.
Q: What are the advantages of using purine salts over free base alkylation?
A: Using pre-formed purine salts, such as the tetrabutylammonium or benzyltriethylammonium salts described in the patent, significantly enhances nucleophilicity and solubility in organic solvents. This allows the alkylation to proceed under much milder conditions compared to traditional methods that require strong, hazardous bases, thereby improving reaction control and reducing side products.
Q: How is stereochemical integrity maintained during the coupling reaction?
A: The process utilizes optically active cyclobutane precursors where the leaving group is cis to the adjacent protected hydroxymethyl group. The nucleophilic substitution at the N9 position of the purine proceeds with inversion of configuration, ensuring the correct [1R-(1α,2β,3α)] stereochemistry required for antiviral activity in the final product.
Q: Which leaving groups provide the best balance of reactivity and stability?
A: The patent highlights perfluoroalkanesulfonyloxy groups, specifically trifluoromethanesulfonyloxy (triflate), and nitro-substituted benzenesulfonyloxy groups, such as p-nitrobenzenesulfonyloxy (nosylate). Triflates offer high reactivity allowing reactions at 0°C to 30°C, while nosylates provide a stable alternative that reacts efficiently at slightly elevated temperatures in acetonitrile.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Carbocyclic Purine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthetic routes in the development of life-saving antiviral medications. Our team of expert chemists has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the rigorous demands of global pharmaceutical clients. We are committed to delivering high-purity intermediates that adhere to stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our facility is designed to handle complex organic syntheses safely and efficiently, making us an ideal partner for your long-term supply needs.
We invite you to collaborate with us to leverage this advanced patent technology for your next project. Our technical sales team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality standards. Please contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you optimize your supply chain and bring your antiviral drug candidates to market faster and more cost-effectively.