Scalable Manufacturing of Asciminib Intermediates via Optimized Palladium Catalysis and Amide Coupling
The pharmaceutical industry continuously seeks robust manufacturing routes for complex kinase inhibitors, particularly for oncology applications where supply chain reliability is paramount. Patent CN114096529A, published on February 25, 2022, introduces a significantly improved chemical process for the synthesis of N-[4-(chlorodifluoromethoxy)phenyl]-6-[(3R)-3-hydroxypyrrolidin-1-yl]-5-(1H-pyrazol-5-yl)pyridine-3-carboxamide, widely known as Asciminib. This compound is a potent BCR-ABL tyrosine-kinase inhibitor, representing a critical therapeutic modality for treating chronic myeloid leukemia. The disclosed methodology addresses long-standing challenges in the prior art, specifically focusing on enhancing reaction conversion, simplifying purification protocols, and ensuring reproducibility at commercial production scales. By optimizing key catalytic steps and reagent selections, this patent offers a pathway that minimizes waste generation while maximizing the purity of the final active pharmaceutical ingredient (API) intermediate.
For research and development directors evaluating process viability, the technical nuances of this invention are particularly compelling. The process leverages advanced palladium catalysis to construct the core biaryl scaffold with exceptional efficiency. Unlike earlier methods that struggled with incomplete conversions and difficult-to-remove impurities during scale-up, the new protocol utilizes a specific ligand-metal complex that maintains high activity even in large-volume reactors. This ensures that the critical carbon-carbon bond formation proceeds to near completion, drastically reducing the burden on downstream purification units. Furthermore, the strategic selection of bases and solvents facilitates phase separation and crystallization, which are often the bottlenecks in fine chemical manufacturing. The result is a streamlined workflow that aligns perfectly with the rigorous quality standards required for high-purity pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art processes, such as those described in WO 2013/171639 A1, relied on less efficient catalytic systems and more cumbersome coupling strategies that proved problematic upon scaling. For instance, the conventional Suzuki coupling utilized preformed catalysts like Pd(PPh3)2Cl2 in conjunction with potassium phosphate. While effective on a small laboratory scale, these conditions frequently led to incomplete conversion of starting materials when the reaction volume was increased to several kilograms. This incomplete conversion necessitated extensive chromatographic purification or multiple recrystallizations to remove unreacted halides and boronic acid derivatives, thereby driving up costs and extending lead times. Additionally, the traditional amide coupling approach involved converting a nicotinic acid intermediate using activating agents like EDCI and HOBt. This generated significant amounts of urea byproducts and required the isolation of a carboxylic acid intermediate that was notoriously difficult to crystallize and extract, leading to sustained yield losses and operational inefficiencies in the plant.
The Novel Approach
The innovative strategy presented in CN114096529A fundamentally reengineers these critical steps to overcome the aforementioned bottlenecks. Instead of relying on generic phosphine ligands, the new process employs a specialized palladium catalyst system, specifically PdCl2(dtbpf), which demonstrates superior stability and turnover numbers. This change alone ensures that the Suzuki coupling proceeds with greater than 98% conversion, effectively eliminating the issue of residual starting materials that plagued previous methods. Furthermore, the amide bond formation is reinvented by reacting the methyl ester intermediate directly with 4-(chlorodifluoromethoxy)aniline hydrochloride in the presence of potassium tert-butoxide. This direct aminolysis approach bypasses the need for isolating the unstable nicotinic acid and avoids the use of expensive and waste-generating coupling reagents. The outcome is a cleaner reaction profile that facilitates straightforward filtration and crystallization, significantly enhancing the overall throughput and economic viability of the synthesis.
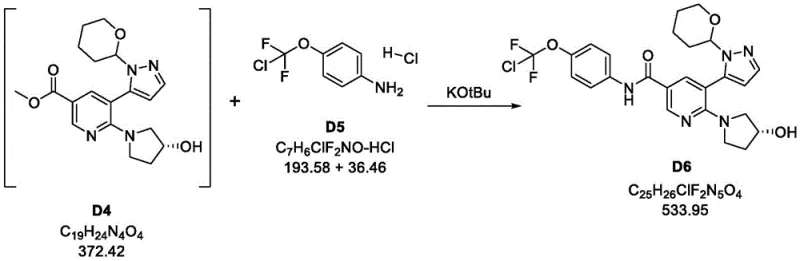
This shift in synthetic logic not only improves the chemical yield but also transforms the physical handling characteristics of the intermediates. By avoiding the formation of sticky oils or difficult-to-filter solids common in the old EDCI-mediated process, the new method ensures smooth operation in standard stainless steel reactors. The ability to perform solvent swaps and crystallizations with high recovery rates means that the process is inherently more robust against variations in raw material quality. For procurement managers, this translates to a more predictable supply of reliable pharmaceutical intermediates supplier outputs, as the risk of batch failure due to purification issues is substantially mitigated. The process design inherently supports cost reduction in pharmaceutical intermediates manufacturing by reducing the number of unit operations and the consumption of auxiliary chemicals.
Mechanistic Insights into Pd-Catalyzed Cross-Coupling and Deprotection
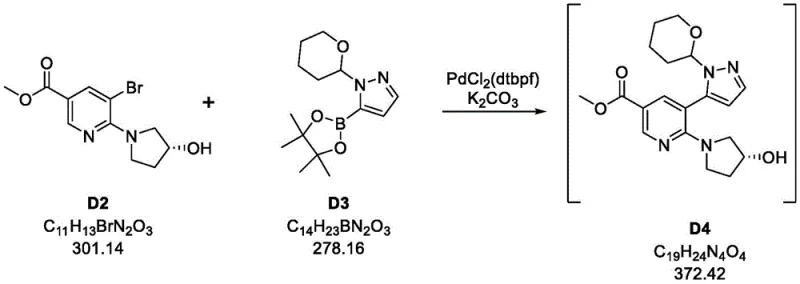
The core of this synthetic advancement lies in the meticulous optimization of the palladium-catalyzed cross-coupling mechanism. The reaction between the bromo-substituted nicotinate (Compound D2) and the pyrazole boronic ester (Compound D3) is facilitated by the PdCl2(dtbpf) catalyst. The dtbpf ligand (1,1'-bis(di-tert-butylphosphino)ferrocene) creates a sterically bulky and electron-rich environment around the palladium center. This specific electronic configuration accelerates the oxidative addition of the aryl bromide and facilitates the subsequent transmetallation with the boron species. Crucially, the use of potassium carbonate as the base in a biphasic toluene-water system promotes the formation of the reactive boronate species while maintaining the solubility of the organic intermediates. This mechanistic precision prevents the formation of homocoupling byproducts and ensures that the biaryl linkage is formed with high regioselectivity. The result is an intermediate (Compound D4) that is obtained in approximately 95% yield, setting a high-purity foundation for the subsequent steps.
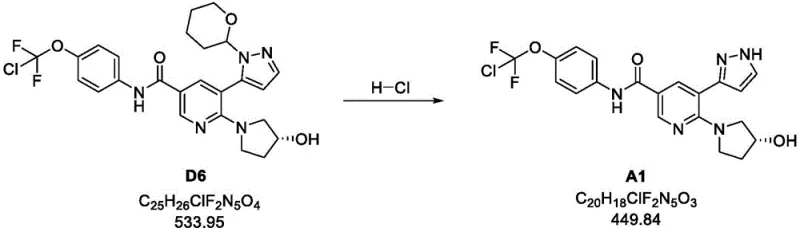
Following the coupling, the deprotection mechanism is equally refined to ensure the integrity of the sensitive pyrazole and pyrrolidine moieties. The removal of the tetrahydropyranyl (THP) protecting group from Compound D6 is achieved using aqueous hydrochloric acid in methanol. This acid-catalyzed hydrolysis proceeds through the protonation of the acetal oxygen, followed by the elimination of the dihydropyran side product. The patent highlights the importance of controlled pH adjustment during the workup; after the initial acidic cleavage, the mixture is carefully neutralized to a specific pH range (3.0-3.5) before final basification. This precise pH control is critical for inducing the crystallization of the final product (Compound A1) while keeping soluble impurities in the mother liquor. The seeding technique employed during this stage further directs the polymorphic form and particle size distribution, which are essential parameters for downstream formulation. This level of control over the solid-state properties underscores the process's suitability for commercial scale-up of complex pharmaceutical intermediates.
How to Synthesize Asciminib Intermediate Efficiently
The synthesis of this high-value kinase inhibitor intermediate is structured around three pivotal transformations that have been rigorously optimized for industrial application. The process begins with the construction of the heterobiaryl core, followed by the installation of the aniline side chain, and concludes with the global deprotection to reveal the active pharmacophore. Each step has been designed to maximize atom economy and minimize the environmental footprint, adhering to green chemistry principles where possible. The detailed operational parameters, including specific temperature ramps, addition rates, and stoichiometric ratios, are critical for reproducing the high yields reported in the patent examples. Operators must pay close attention to the water content in the Suzuki coupling and the exotherm management during the amide formation to ensure safety and consistency. For a comprehensive guide on executing these reactions with GMP-level precision, please refer to the standardized protocol below.
- Perform Suzuki coupling of bromo-nicotinate (D2) with pyrazole boronic ester (D3) using PdCl2(dtbpf) and K2CO3 in toluene/water to yield intermediate D4.
- React intermediate D4 with 4-(chlorodifluoromethoxy)aniline hydrochloride (D5) using potassium tert-butoxide in methyltetrahydrofuran to form protected amide D6.
- Deprotect the tetrahydropyranyl group of D6 using aqueous HCl in methanol, followed by neutralization and crystallization to obtain the final free base A1.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain perspective, the adoption of this novel synthetic route offers transformative benefits that extend far beyond simple yield improvements. The elimination of problematic purification steps and the use of robust, commercially available catalysts create a manufacturing profile that is highly resilient to market fluctuations. By removing the dependency on difficult-to-handle intermediates and expensive coupling reagents, the process inherently lowers the cost of goods sold (COGS). This efficiency gain is passed down the value chain, allowing for more competitive pricing structures without compromising on quality. Moreover, the simplified workflow reduces the total cycle time per batch, enabling manufacturers to respond more agilely to demand surges. This agility is crucial for maintaining reducing lead time for high-purity pharmaceutical intermediates in a volatile global market.
- Cost Reduction in Manufacturing: The most significant economic driver of this process is the replacement of the EDCI/HOBt coupling system with a direct aminolysis using potassium tert-butoxide. Carbodiimide reagents are not only costly but also generate stoichiometric amounts of urea waste that require disposal. By switching to a base-mediated amidation, the process eliminates this waste stream entirely, resulting in substantial cost savings in both raw material procurement and waste treatment. Additionally, the high conversion rate of the Suzuki coupling means that precious palladium catalysts are utilized more efficiently, reducing the effective catalyst loading required per kilogram of product. These cumulative efficiencies drive down the overall production cost, making the final API more accessible.
- Enhanced Supply Chain Reliability: The robustness of the new method significantly de-risks the supply chain. In the prior art, the difficulty in crystallizing the nicotinic acid intermediate often led to batch delays and extended processing times as engineers struggled to isolate the material. The new process generates intermediates that crystallize readily and predictably, ensuring consistent batch-to-batch performance. This reliability allows supply chain planners to forecast production schedules with greater accuracy, minimizing the risk of stockouts. Furthermore, the reagents used, such as PdCl2(dtbpf) and potassium tert-butoxide, are commodity chemicals with stable supply lines, unlike some specialized activating agents that may face availability constraints. This ensures a continuous flow of materials for commercial scale-up of complex pharmaceutical intermediates.
- Scalability and Environmental Compliance: Scalability is inherent in the design of this process, as evidenced by the successful translation of the Suzuki coupling from gram to multi-kilogram scales without loss of efficiency. The use of common solvents like toluene, methanol, and methyltetrahydrofuran simplifies solvent recovery and recycling operations, aligning with stringent environmental regulations. The reduction in byproduct formation means less chemical waste is sent for incineration or treatment, lowering the facility's environmental impact score. This compliance is increasingly important for multinational corporations aiming to meet sustainability goals. The process demonstrates that high-efficiency synthesis and environmental stewardship are not mutually exclusive, paving the way for greener manufacturing practices in the oncology sector.
Frequently Asked Questions (FAQ)
Understanding the technical specifics of this patented process is essential for stakeholders evaluating its implementation. The following questions address common inquiries regarding catalyst selection, impurity profiles, and operational safety. These answers are derived directly from the experimental data and technical disclosures within CN114096529A, providing a factual basis for decision-making. Whether you are concerned about the removal of heavy metals or the stability of the intermediates, the following insights clarify how this method achieves its superior performance metrics compared to legacy routes.
Q: Why is the PdCl2(dtbpf) catalyst preferred for the Suzuki coupling step in this process?
A: The patent specifies that PdCl2(dtbpf) provides superior conversion rates (>98%) compared to prior art catalysts like Pd(PPh3)2Cl2, which suffered from incomplete conversion at larger scales (e.g., 3kg), ensuring higher purity and yield for the critical biaryl bond formation.
Q: How does the new amide coupling method improve upon the traditional EDCI/HOBt protocol?
A: The novel method utilizes 4-(chlorodifluoromethoxy)aniline hydrochloride directly with potassium tert-butoxide, bypassing the need for difficult-to-crystallize nicotinic acid intermediates and avoiding the generation of urea byproducts associated with carbodiimide coupling reagents, thus simplifying purification.
Q: What are the scalability benefits of the deprotection step described in Example 3?
A: The deprotection uses standard aqueous HCl in methanol followed by a controlled pH adjustment and seeding crystallization. This avoids hazardous reagents and allows for robust isolation of the product with a yield of approximately 76%, making it highly suitable for multi-kilogram production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Asciminib Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent literature to commercial reality requires deep technical expertise and state-of-the-art infrastructure. As a leading CDMO partner, we possess the capability to translate the innovative pathways described in CN114096529A into reliable, large-scale production. Our facilities are equipped to handle the specific requirements of palladium-catalyzed reactions, including dedicated containment for potent compounds and advanced filtration systems for catalyst removal. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with unwavering consistency. Our commitment to quality is underpinned by stringent purity specifications and rigorous QC labs that verify every batch against the highest industry standards, guaranteeing that the Asciminib intermediates we deliver are ready for immediate downstream processing.
We invite potential partners to engage with our technical team to explore how this optimized process can enhance your supply chain resilience. By leveraging our manufacturing prowess, you can secure a steady source of high-quality intermediates while benefiting from the cost efficiencies inherent in this novel route. We encourage you to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. Our technical procurement team is ready to provide specific COA data and route feasibility assessments to demonstrate how we can support your long-term strategic goals in the oncology therapeutic area.