Advanced Quality Control for Sitagliptin Intermediates: Ensuring Global Compliance and Scalability
The pharmaceutical industry continuously faces the challenge of maintaining stringent purity standards for active pharmaceutical ingredients (APIs), particularly for high-volume drugs like Sitagliptin phosphate. Patent CN109824546B introduces a critical advancement in the quality control of Sitagliptin synthesis by focusing on a specific condensation impurity found in the key intermediate, BOC-(R)-3-amino-4-(2,4,5-trifluorophenyl) butyric acid. This innovation addresses a significant bottleneck where traditional methods often struggle to keep single impurity levels below the 0.10% threshold mandated by the European and United States Pharmacopoeias. By providing a reliable method to synthesize and control this specific condensation impurity, the patent offers a robust pathway for manufacturers to ensure the final bulk drug meets all regulatory requirements without resorting to yield-damaging purification techniques. This technical breakthrough is essential for any reliable pharmaceutical intermediates supplier aiming to support the global production of Type II diabetes medications with consistent quality and safety profiles.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Sitagliptin phosphate has been plagued by the formation of difficult-to-remove impurities that emerge during the condensation steps of the intermediate production. Conventional approaches often rely on post-synthesis recrystallization to purify the final product, a method that is inherently inefficient and costly for large-scale industrial operations. When the content of specific condensation impurities in the intermediate exceeds critical thresholds, it leads to the generation of Impurity A in the final API, which frequently surpasses the strict 0.10% limit. This forces manufacturers to implement additional purification cycles, which drastically reduce overall yield and increase the consumption of solvents and raw materials. Furthermore, the reliance on end-product testing rather than intermediate control creates a reactive quality assurance environment, where batches may be rejected late in the production cycle, causing significant supply chain disruptions and financial losses for pharmaceutical companies.
The Novel Approach
The methodology outlined in patent CN109824546B shifts the quality control paradigm from reactive final testing to proactive intermediate management. By synthesizing a high-purity reference standard of the BOC-(R)-3-amino-4-(2,4,5-trifluorophenyl) butyric acid condensation impurity, manufacturers can accurately monitor its levels during the intermediate synthesis phase. The patent demonstrates that keeping this specific impurity below 0.20% in the intermediate effectively prevents the formation of unacceptable levels of Impurity A in the final Sitagliptin product. This approach simplifies the overall manufacturing process by eliminating the need for aggressive final recrystallization, thereby preserving yield and reducing operational complexity. The ability to detect and quantify this impurity using liquid chromatography with a specific reference standard allows for precise process adjustments, ensuring that every batch meets the rigorous standards required for commercial drug production.
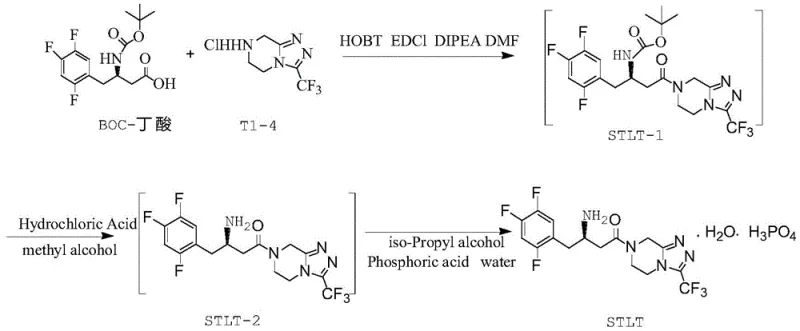
Mechanistic Insights into Condensation Impurity Formation
The formation of the condensation impurity is a direct result of the coupling reaction between the protected amino acid and the free amino acid during the intermediate synthesis. Understanding the mechanistic pathway is crucial for R&D directors focused on process optimization and impurity profiling. The reaction typically involves the activation of the carboxylic acid group of the BOC-protected species using carbodiimide coupling agents, which then reacts with the amine group of the free acid. However, under certain conditions, a side reaction occurs where two molecules of the amino acid derivative condense, forming a dimer-like structure that persists through subsequent synthetic steps. This specific condensation byproduct is structurally similar to the desired intermediate, making it challenging to separate without specialized chromatographic techniques. The patent details the precise chemical structure of this impurity, providing a clear target for analytical method development and process control strategies.
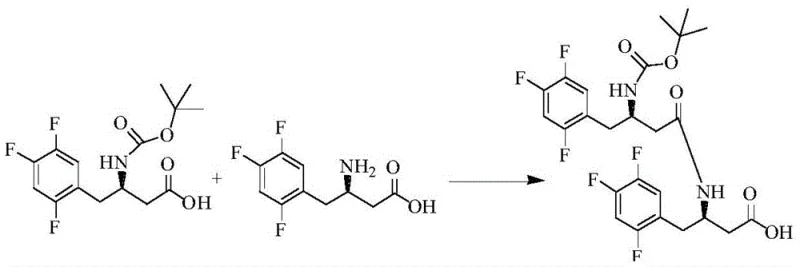
Controlling the stoichiometry and reaction conditions is paramount to minimizing the generation of this unwanted byproduct. The patent specifies the use of coupling agents like 1-Hydroxybenzotriazole (HOBT) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC-HCl) in the presence of an organic base such as triethylamine. The molar ratios of the reactants are carefully optimized, with a preferred ratio of BOC-protected acid to free acid ranging from 1:1.0 to 1:1.1 to drive the reaction towards the desired product while suppressing dimerization. Temperature control between 20-30°C is also critical, as higher temperatures can accelerate side reactions leading to increased impurity levels. By adhering to these specific mechanistic parameters, manufacturers can significantly reduce the burden on downstream purification processes, ensuring a cleaner reaction profile and a more robust manufacturing process.
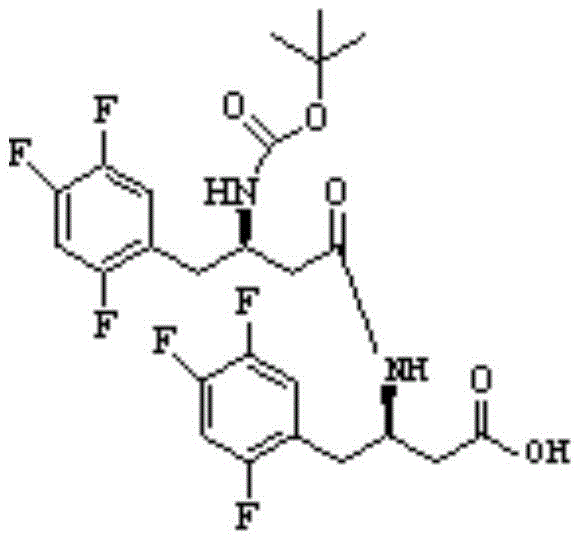
How to Synthesize BOC-(R)-3-amino-4-(2,4,5-trifluorophenyl) butyric acid Efficiently
To achieve high-purity intermediates suitable for Sitagliptin production, it is essential to follow a standardized synthesis protocol that prioritizes impurity control from the outset. The detailed procedure involves dissolving the starting materials in an anhydrous organic solvent like acetonitrile and adding the coupling reagents under controlled temperature conditions. The reaction mixture is then monitored using liquid chromatography to ensure the condensation impurity remains within the specified limits before proceeding to the next step. For a comprehensive guide on the exact reagent quantities, reaction times, and purification techniques required to replicate this process at scale, please refer to the standardized synthesis steps provided below.
- Dissolve BOC-(R)-3-amino-4-(2,4,5-trifluorophenyl) butanoic acid and (R)-3-amino-4-(2,4,5-trifluorophenyl) butanoic acid in an organic solvent such as acetonitrile.
- Add coupling agents like HOBT and EDC-HCl along with an organic base such as triethylamine while maintaining the temperature between 20-30°C.
- Purify the resulting crude product using silica gel column chromatography to isolate the high-purity condensation impurity for analytical standards.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the implementation of this impurity control strategy offers substantial operational benefits that translate directly to cost efficiency and reliability. By preventing the formation of critical impurities early in the synthesis, the overall process becomes more streamlined, reducing the need for expensive and time-consuming purification steps in the final stages. This efficiency gain means that raw materials are utilized more effectively, leading to a significant reduction in the cost of goods sold without compromising on the quality of the final API. Additionally, a more predictable synthesis process enhances supply chain stability, as the risk of batch failure due to purity issues is markedly decreased. This reliability is crucial for maintaining continuous production schedules and meeting the demanding delivery timelines of global pharmaceutical clients.
- Cost Reduction in Manufacturing: The elimination of complex recrystallization steps for the final product results in substantial cost savings by reducing solvent consumption and labor hours. Since the impurity is controlled at the intermediate stage, the yield of the final Sitagliptin phosphate is preserved, maximizing the output from each batch of raw materials. This efficiency allows for a more competitive pricing structure for the intermediate, providing a clear economic advantage for manufacturers looking to optimize their production budgets. Furthermore, the reduced need for waste treatment associated with extensive purification processes contributes to lower environmental compliance costs.
- Enhanced Supply Chain Reliability: Implementing strict intermediate control measures ensures a consistent supply of high-quality materials, minimizing the risk of production delays caused by out-of-specification batches. This stability is vital for pharmaceutical companies that operate on tight schedules and cannot afford interruptions in their API supply. By partnering with a supplier that adheres to these rigorous quality standards, procurement teams can secure a dependable source of intermediates that meet all regulatory requirements. This reliability fosters stronger long-term partnerships and reduces the administrative burden associated with managing quality deviations and supplier audits.
- Scalability and Environmental Compliance: The simplified process described in the patent is inherently more scalable, as it avoids the bottlenecks associated with low-yield purification techniques. This scalability allows manufacturers to easily ramp up production to meet increasing market demand for diabetes medications without significant capital investment in new equipment. Moreover, the reduction in solvent usage and waste generation aligns with modern environmental sustainability goals, making the process more attractive to companies focused on green chemistry initiatives. This compliance with environmental standards not only mitigates regulatory risk but also enhances the corporate social responsibility profile of the manufacturing entity.
Frequently Asked Questions (FAQ)
The following questions address common concerns regarding the implementation of this impurity control method and its impact on the overall synthesis of Sitagliptin phosphate. These answers are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and relevance for technical decision-makers. Understanding these details is essential for evaluating the feasibility of adopting this approach in your own manufacturing processes and for assessing the potential benefits for your supply chain.
Q: Why is controlling condensation impurities critical in Sitagliptin synthesis?
A: Controlling these impurities at the intermediate stage prevents the formation of Impurity A in the final API, ensuring compliance with pharmacopoeia limits of less than 0.10% single impurity without costly recrystallization.
Q: What reagents are typically used for this condensation reaction?
A: The process utilizes standard peptide coupling reagents such as 1-Hydroxybenzotriazole (HOBT) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC-HCl) in the presence of an organic base.
Q: How does this method improve industrial production efficiency?
A: By monitoring and limiting the condensation impurity to below 0.20% in the intermediate, manufacturers can avoid complex purification steps in the final stage, significantly reducing production time and material loss.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Sitagliptin Intermediate Supplier
At NINGBO INNO PHARMCHEM, we understand the critical importance of impurity control in the production of high-value pharmaceutical intermediates like those used for Sitagliptin phosphate. Our team of experts possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that every batch meets stringent purity specifications. We utilize rigorous QC labs to monitor critical parameters, guaranteeing that our products consistently comply with international pharmacopoeia standards. Our commitment to quality and technical excellence makes us the ideal partner for pharmaceutical companies seeking to optimize their supply chain and ensure the safety and efficacy of their final drug products.
We invite you to contact our technical procurement team to discuss how our advanced manufacturing capabilities can support your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain valuable insights into how our processes can reduce your overall production costs while maintaining the highest quality standards. We are ready to provide specific COA data and route feasibility assessments to help you make informed decisions about your supply chain strategy. Let us help you engineer a more efficient and reliable path to market for your pharmaceutical products.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →