Advanced Synthesis of Gemcitabine Hydrochloride via 1,6-Dehydro-beta-D-Glucose for Commercial Scale
The pharmaceutical industry continuously seeks robust synthetic pathways for critical antiviral agents, and Patent CN1228342C presents a significant advancement in the preparation of 2'-deoxy-2',2'-difluoro-beta-nucleoside derivatives, specifically Gemcitabine Hydrochloride. This patent discloses a novel methodology utilizing 1,6-dehydro-beta-D-glucose as the primary raw material, which undergoes a series of oxidation and fluorination steps to yield the key intermediate 2'-deoxy-2',2'-difluoro-D-ribofuranose. Unlike traditional routes that rely on D-Glyceraldehyde and suffer from stereoselectivity issues, this approach leverages the rigid structural integrity of the glucose derivative to ensure high optical purity throughout the synthesis. The process is characterized by its simplicity, high output rate, and exceptional suitability for scale industrial production, addressing the growing global demand for high-quality oncology therapeutics. By establishing a reliable supply chain for this complex molecule, manufacturers can mitigate risks associated with raw material volatility and ensure consistent availability for downstream drug formulation.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of beta-1-(2'-deoxy-2',2'-difluoro-D-ribofuranosyl)-4-aminopyrimidine-2-keto hydrochloride has predominantly relied on D-Glyceraldehyde as the starting chiral pool material, a method extensively documented in literature such as Hertel et al. and Chou et al. However, this conventional pathway is fundamentally flawed due to its reliance on the Reformatsky reaction, which is inherently non-stereoselective and fails to adequately control the stereochemistry at the critical 2'-position of the ribofuranose ring. Furthermore, the traditional process necessitates the use of low-boiling, anhydrous, and oxygen-free ethers as solvents, which introduces significant safety hazards and operational complexities in a manufacturing environment. The requirement for extensive solvent crystallization to separate isomers not only drives up processing costs but also results in substantially lower overall yields, making the process economically inefficient for large-scale operations. These technical bottlenecks create substantial barriers for procurement managers seeking cost reduction in API manufacturing, as the poor controllability of the reaction pilot process leads to inconsistent batch quality and extended production timelines.
The Novel Approach
In stark contrast to legacy methods, the novel approach detailed in the patent utilizes 1,6-dehydro-beta-D-glucose, a stable and readily available carbohydrate derivative, to bypass the stereoselectivity challenges entirely. This innovative route allows for the fine control of stereoselective reactions throughout the build-up process, ensuring the efficient acquisition of the chiral compound 2'-deoxy-2',2'-difluoro-beta-nucleic cytidine without the need for difficult isomer separations. The method streamlines the synthesis into distinct, manageable steps involving oxidation, fluorination, and condensation, each optimized for high transformation efficiency and minimal byproduct formation. By eliminating the need for hazardous ether solvents and complex crystallization steps, this new pathway drastically simplifies the operational workflow, thereby enhancing supply chain reliability and reducing the environmental footprint of the manufacturing process. This shift represents a paradigm change for supply chain heads, offering a scalable solution that ensures continuity of supply while adhering to stringent environmental compliance standards required in modern chemical production facilities.
Mechanistic Insights into Stereoselective Fluorination and Oxidative Cleavage
The core of this synthetic breakthrough lies in the precise manipulation of the carbohydrate scaffold, where the rigid structure of 1,6-dehydro-beta-D-glucose serves as a template for introducing the difluoro motif with high fidelity. The mechanism begins with the selective protection of hydroxyl groups using trimethylchlorosilane, followed by oxidation with Dess-Martin reagent to generate the 3-ketone intermediate, a step chosen for its mild conditions and high conversion rates. Subsequent fluorination employs a synergistic mixture of DAST and DMPU-HF, which acts to replace the carbonyl oxygen with fluorine atoms while maintaining the stereochemical integrity of the adjacent chiral centers. This specific reagent combination is critical, as it shortens reaction times and prevents the degradation of the sensitive sugar backbone that often occurs with harsher fluorinating agents. The final oxidative cleavage using sodium periodate selectively breaks the carbon-carbon bond to form the furanose ring, completing the construction of the nucleoside core with the correct beta-configuration essential for biological activity.
Controlling the impurity profile in the synthesis of high-purity pharmaceutical intermediates is paramount, and this patent addresses this through specific protection and deprotection strategies that minimize side reactions. The use of TMS blocking groups allows for selective reactivity, ensuring that only the desired hydroxyl groups participate in the oxidation and fluorination steps, thereby reducing the formation of regio-isomers. Furthermore, the hydrolysis steps are conducted under controlled acidic conditions to prevent the epimerization of the chiral centers, which is a common source of impurities in nucleoside synthesis. The purification protocols described, including simple solvent extraction and filtration, are designed to remove inorganic salts and organic byproducts without requiring chromatographic separation, which is often a bottleneck in commercial scale-up of complex pharmaceutical intermediates. This rigorous approach to impurity control ensures that the final product meets the stringent purity specifications required by regulatory bodies, providing R&D directors with confidence in the quality and reproducibility of the synthetic route.
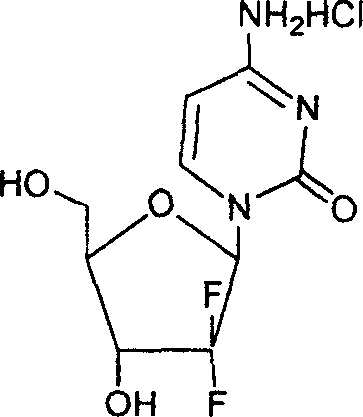
How to Synthesize 2'-Deoxy-2',2'-difluoro-beta-Nucleoside Efficiently
The synthesis of this critical oncology intermediate requires a disciplined approach to reaction conditions and reagent selection to maximize yield and purity while maintaining operational safety. The process begins with the silylation of 1,6-dehydro-beta-D-glucose in anhydrous methylene chloride, followed by oxidation and fluorination steps that must be carefully monitored to prevent over-reaction or decomposition. Detailed standard operating procedures for each transformation, including specific molar ratios, temperature ranges, and work-up protocols, are essential for replicating the high success rates reported in the patent data. For technical teams looking to implement this route, understanding the nuances of the fluorination step, particularly the synergy between DAST and DMPU-HF, is crucial for achieving the desired transformation efficiency. The following guide outlines the standardized synthesis steps derived from the patent claims to facilitate technology transfer and process validation.
- Protect hydroxyl groups of 1,6-dehydro-beta-D-glucose using trimethylchlorosilane to form silyl ethers.
- Oxidize the protected intermediate using Dess-Martin reagent to introduce the ketone functionality at the 3-position.
- Perform fluorination using DAST and DMPU-HF mixture followed by hydrolysis and oxidative cleavage to form the ribofuranose core.
Commercial Advantages for Procurement and Supply Chain Teams
Adopting this novel synthetic route offers profound commercial advantages for procurement and supply chain teams by fundamentally altering the cost structure and risk profile of Gemcitabine production. The elimination of the Reformatsky reaction and the associated complex isomer separation steps translates directly into significant cost savings, as it reduces the consumption of expensive reagents and minimizes waste generation. Furthermore, the use of stable, non-hazardous solvents like methylene chloride and acetonitrile simplifies regulatory compliance and reduces the need for specialized containment equipment, lowering capital expenditure requirements for manufacturing facilities. This streamlined process enhances supply chain reliability by reducing the number of unit operations and potential failure points, ensuring a more consistent flow of high-purity intermediates to downstream formulation sites. For organizations focused on cost reduction in API manufacturing, this technology provides a clear pathway to margin improvement without compromising on the quality or safety of the final therapeutic product.
- Cost Reduction in Manufacturing: The new method eliminates the need for expensive transition metal catalysts and complex chromatographic purification steps, which are major cost drivers in traditional nucleoside synthesis. By utilizing readily available starting materials like 1,6-dehydro-beta-D-glucose and optimizing reagent stoichiometry, the overall material cost is substantially reduced, allowing for more competitive pricing in the global market. Additionally, the high transformation efficiency of the fluorination step minimizes raw material waste, further contributing to the economic viability of the process on a commercial scale. These factors combine to create a robust economic model that supports long-term sustainability and profitability for manufacturers adopting this technology.
- Enhanced Supply Chain Reliability: The robustness of the synthetic route, characterized by its tolerance to variations in reaction conditions and the stability of intermediates, ensures a consistent supply of critical materials. The avoidance of low-boiling, hazardous solvents reduces the risk of supply disruptions related to solvent availability or transportation regulations, thereby stabilizing the production schedule. This reliability is crucial for reducing lead time for high-purity pharmaceutical intermediates, allowing manufacturers to respond more agilely to fluctuations in market demand. By securing a stable and predictable supply chain, companies can better manage inventory levels and reduce the risk of stockouts that could impact patient access to essential medications.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing unit operations that are easily transferred from laboratory to pilot and commercial scales without significant re-engineering. The use of environmentally benign reagents and the generation of minimal hazardous waste align with modern green chemistry principles, facilitating easier permitting and regulatory approval in diverse jurisdictions. This environmental compliance not only mitigates regulatory risk but also enhances the corporate social responsibility profile of the manufacturer, appealing to ethically conscious partners and investors. The ability to scale from 100 kgs to 100 MT annual commercial production with consistent quality makes this route an ideal candidate for meeting the growing global demand for antiviral and anticancer therapies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthetic method, based on the detailed specifications and experimental data provided in the patent documentation. These insights are intended to clarify the operational benefits and technical feasibility of the route for stakeholders evaluating its adoption for commercial production. Understanding these nuances is essential for making informed decisions about process development and supply chain integration.
Q: What is the primary advantage of using 1,6-dehydro-beta-D-glucose over D-Glyceraldehyde?
A: Using 1,6-dehydro-beta-D-glucose allows for better control of stereoselectivity during the formation of the chiral intermediate, avoiding the non-stereoselective Reformatsky reaction associated with D-Glyceraldehyde routes.
Q: How does the new fluorination method improve yield?
A: The synergistic use of DAST and DMPU-HF mixture significantly shortens reaction time and increases transformation efficiency compared to using DAST alone.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the method utilizes stable starting materials and avoids complex solvent crystallization steps, making it highly suitable for repetition and scale preparation.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Gemcitabine Hydrochloride Supplier
NINGBO INNO PHARMCHEM stands at the forefront of chemical innovation, leveraging advanced synthetic methodologies like the one described in Patent CN1228342C to deliver superior value to our global partners. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and reliability. We are committed to maintaining stringent purity specifications and operating rigorous QC labs to guarantee that every batch of Gemcitabine Hydrochloride meets the highest international standards. By partnering with us, you gain access to a wealth of technical expertise and a robust manufacturing infrastructure capable of handling complex chemical transformations with efficiency and safety.
We invite you to collaborate with our technical procurement team to explore how this advanced synthesis route can optimize your supply chain and reduce overall manufacturing costs. Request a Customized Cost-Saving Analysis today to understand the specific economic benefits applicable to your production volume and requirements. Our experts are ready to provide specific COA data and route feasibility assessments to support your decision-making process and ensure a seamless transition to this superior manufacturing technology. Let us help you secure a competitive edge in the market with our reliable supply of high-quality pharmaceutical intermediates.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →