Advanced Stereoselective Synthesis of Gemcitabine Hydrochloride for Commercial Scale-up
The pharmaceutical landscape for oncology treatments continues to evolve, with a persistent demand for efficient manufacturing routes for critical nucleoside analogs. Patent CN101381387B introduces a significant methodological advancement in the stereoselective preparation of 2'-deoxy-2',2'-difluoro-β-cytidine hydrochloride, widely known as Gemcitabine Hydrochloride. This compound serves as a pivotal pyrimidine antimetabolite, demonstrating unique anti-tumor activity against solid tumors including pancreatic and non-small-cell lung cancer. The disclosed technology addresses long-standing challenges in nucleoside synthesis by leveraging kinetic resolution during the halogenation phase, thereby circumventing the need for cryogenic reaction conditions that have historically plagued industrial scale-up efforts. By optimizing the conversion of ribofuranose precursors, this approach offers a robust pathway for producing high-purity pharmaceutical intermediates suitable for global supply chains. 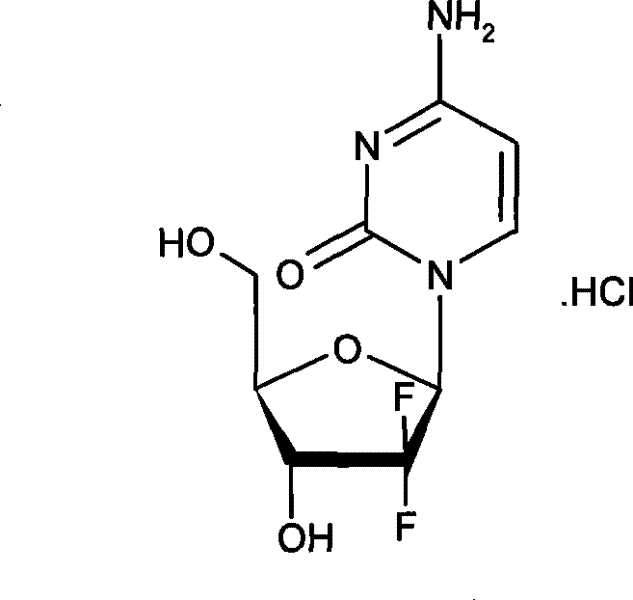
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Gemcitabine has relied heavily on activation strategies that impose severe operational constraints on manufacturing facilities. Conventional pathways, such as those described in earlier literature, often necessitate the activation of the 1-position hydroxyl group of 2',2'-difluororibofuranose to create an active intermediate. A predominant limitation in these legacy methods is the stringent requirement for extremely low reaction temperatures, often dropping below minus 80 degrees Celsius, to achieve acceptable stereoselectivity ratios between alpha and beta isomers. Such cryogenic conditions are not only energy-intensive but also introduce significant engineering bottlenecks when attempting to transition from laboratory bench scale to multi-ton commercial production. Furthermore, alternative routes utilizing reagents like diphenyl phosphoryl chloride involve exorbitant raw material costs and complex workup procedures to remove phosphorus-containing byproducts. The necessity to separate alpha and beta isomers at the intermediate stage further exacerbates yield losses and increases the consumption of organic solvents, rendering these traditional methods economically inefficient for high-volume API manufacturing. 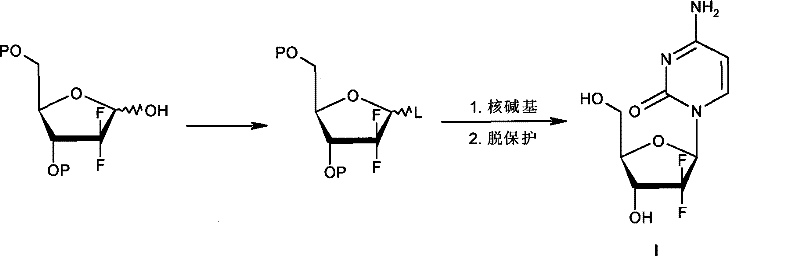
The Novel Approach
The methodology disclosed in the patent data presents a paradigm shift by exploiting the inherent kinetic differences between stereoisomers during nucleophilic substitution. Instead of relying on thermodynamic control via extreme cooling, this novel approach utilizes a halogenation reaction where the beta-configured methanesulfonate precursor reacts at a velocity approximately twofold that of its alpha-configured counterpart. This kinetic disparity allows for the selective conversion of the beta-isomer into the desired alpha-halide while leaving the alpha-methanesulfonate largely unreacted in the same vessel. Crucially, the process eliminates the need to isolate the intermediate alpha-halide from the unreacted starting material. The resulting mixture, containing both the newly formed alpha-halide and the residual alpha-methanesulfonate, is directly subjected to the subsequent glycosylation step with silylated cytosine. This telescoping of steps not only simplifies the operational workflow but also drastically reduces the physical footprint and equipment time required for production, marking a substantial improvement over the fragmented unit operations of the past.
Mechanistic Insights into Kinetic Halogenation and Glycosylation
The core of this synthetic innovation lies in the precise manipulation of reaction kinetics during the halogenation phase using sodium halide and a phase-transfer catalyst. When the mixture of alpha and beta 1-methanesulfonates ribofuranose is treated with sodium bromide in a solvent system such as tetrahydrofuran, the nucleophilic attack occurs preferentially at the beta-anomeric center. This selectivity is driven by the steric and electronic environment of the sugar ring, where the beta-leaving group is more accessible to the halide ion. As the reaction progresses, monitored via HPLC, the beta-methanesulfonate is consumed and converted into the alpha-halide, while the alpha-methanesulfonate remains kinetically inert under these specific conditions. The result is a reaction mixture enriched with the desired alpha-halide stereoisomer, which is the requisite precursor for forming the beta-nucleoside in the subsequent coupling step. This mechanism effectively acts as a chemical filter, enriching the stereochemical purity of the intermediate without the need for physical separation techniques like chromatography or fractional crystallization. 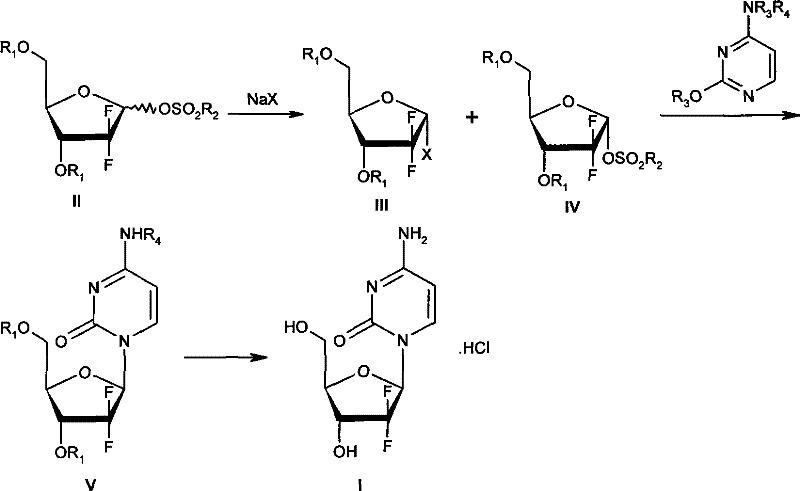
Following the kinetic halogenation, the crude mixture undergoes a glycosylation reaction with silylated cytosine, a process that demands careful control of temperature and solvent composition to maintain stereochemical integrity. The reaction is typically conducted in a mixed solvent system comprising high-boiling ethers and alkanes, such as phenyl ether and normal heptane, at temperatures ranging from 90 to 135 degrees Celsius. Under these thermal conditions, the alpha-halide intermediate reacts with the silylated base to form the N-glycosidic bond, predominantly yielding the beta-nucleoside configuration due to the neighboring group participation or specific transition state geometry. The unreacted alpha-methanesulfonate present in the mixture does not interfere significantly with this coupling, allowing the reaction to proceed to high conversion rates. Subsequent deprotection using saturated ammonia in methanol removes the benzoyl protecting groups from the sugar moiety, and final salt formation with hydrochloric acid yields the target Gemcitabine Hydrochloride with a purity exceeding 99 percent as verified by HPLC analysis. This mechanistic pathway ensures that impurity profiles are tightly controlled, meeting the stringent specifications required for oncology therapeutics.
How to Synthesize Gemcitabine Hydrochloride Efficiently
- React 1-methanesulfonates ribofuranose mixture with sodium halide and phase-transfer catalyst to selectively convert beta-isomer to alpha-halide.
- Couple the resulting crude mixture directly with silylated cytosine in a high-boiling solvent system at 90-135°C.
- Perform deprotection using saturated ammonia methanol followed by salt formation with hydrochloric acid to crystallize the final product.
The implementation of this synthesis route requires a disciplined approach to reaction monitoring and reagent stoichiometry to maximize the benefits of the kinetic resolution. Operators must ensure that the halogenation step is allowed to proceed until HPLC analysis confirms the substantial conversion of the beta-methanesulfonate, typically requiring reaction times around 48 hours depending on the specific solvent and catalyst loading. The detailed standardized synthesis steps, including exact molar ratios of sodium bromide to substrate and specific temperature ramps for the glycosylation phase, are critical for reproducibility. For a comprehensive breakdown of the operational parameters and safety protocols required to execute this chemistry at scale, please refer to the technical guide below which outlines the precise sequence of unit operations.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic procurement perspective, the adoption of this synthetic route offers compelling advantages that directly impact the cost of goods sold and supply chain resilience. The elimination of intermediate separation steps translates to a significant reduction in processing time and solvent consumption, which are major cost drivers in fine chemical manufacturing. By avoiding the need for cryogenic cooling infrastructure, facilities can utilize standard reactor trains, thereby lowering capital expenditure requirements and increasing overall equipment effectiveness. The use of commodity chemicals like sodium bromide and tetrahydrofuran, rather than specialized and expensive phosphorylating agents, further insulates the supply chain from raw material price volatility. These factors combine to create a manufacturing process that is not only economically superior but also more robust against disruptions, ensuring a steady flow of high-quality intermediates to downstream API producers.
- Cost Reduction in Manufacturing: The economic benefits of this process are derived primarily from the telescoping of reaction steps and the removal of purification bottlenecks. By processing the crude halogenation mixture directly into the glycosylation reactor, the manufacturer eliminates the costs associated with isolation, drying, and re-dissolving of intermediates. This reduction in unit operations leads to lower labor costs and decreased energy consumption per kilogram of product. Furthermore, the avoidance of expensive reagents such as diphenyl phosphoryl chloride removes a significant line item from the raw material budget. The cumulative effect of these efficiencies results in a substantially lower production cost structure, allowing for more competitive pricing in the global market for Gemcitabine intermediates without compromising on quality standards.
- Enhanced Supply Chain Reliability: Supply chain continuity is often threatened by the complexity of synthesis routes that rely on hard-to-source reagents or fragile process conditions. This method enhances reliability by utilizing widely available inorganic salts and organic solvents that are standard in the chemical industry. The robustness of the reaction conditions, which do not require extreme low temperatures, reduces the risk of batch failures due to equipment malfunction or cooling system limitations. Additionally, the high conversion rates and selectivity minimize the generation of off-spec material, ensuring that production schedules are met consistently. This predictability is crucial for procurement managers who need to secure long-term supply agreements for critical oncology ingredients, providing confidence in the ability to meet market demand without interruption.
- Scalability and Environmental Compliance: Scaling chemical processes often introduces challenges related to heat transfer and waste management, but this route is inherently designed for industrial feasibility. The exothermic nature of the reactions is manageable within standard reactor configurations, and the reduction in solvent usage per unit of product aligns with green chemistry principles. By minimizing the number of workup and purification stages, the process generates less hazardous waste, simplifying compliance with environmental regulations. The ability to run the reaction at atmospheric pressure and moderate temperatures further enhances safety profiles, making it easier to obtain regulatory approvals for new manufacturing sites. This scalability ensures that the technology can grow with market demand, supporting the transition from pilot plant to multi-ton commercial production seamlessly.
Frequently Asked Questions (FAQ)
Q: How does this method improve stereoselectivity compared to traditional low-temperature routes?
A: Traditional routes often require cryogenic conditions below -80°C to manage stereochemistry. This patent utilizes kinetic control where the beta-methanesulfonate reacts twice as fast as the alpha-isomer, allowing for high selectivity without extreme cooling.
Q: Is intermediate purification required between the halogenation and glycosylation steps?
A: No, a key advantage of this process is the elimination of intermediate separation. The mixture of alpha-halide and unreacted alpha-methanesulfonate is used directly in the subsequent coupling reaction, significantly reducing processing time and solvent waste.
Q: What are the primary cost drivers reduced in this manufacturing process?
A: The process avoids expensive reagents like diphenyl phosphoryl chloride and eliminates costly purification steps for intermediates. Additionally, the use of common solvents like THF and sodium bromide lowers raw material expenses.
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method. These answers are derived directly from the experimental data and technical specifications provided in the patent documentation to ensure accuracy and relevance for industry stakeholders. Understanding these details is essential for R&D teams evaluating the feasibility of technology transfer and for procurement professionals assessing the value proposition of this manufacturing route.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Gemcitabine Hydrochloride Supplier
The technical potential of this stereoselective synthesis route represents a significant opportunity for optimizing the production of oncology intermediates. NINGBO INNO PHARMCHEM, as a seasoned CDMO expert, possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this chemistry to life. Our facilities are equipped with the rigorous QC labs and stringent purity specifications necessary to handle complex nucleoside analogs, ensuring that every batch meets the exacting standards of the pharmaceutical industry. We understand the critical nature of API intermediates in the drug development timeline and are committed to delivering consistent quality and reliability.
We invite you to engage with our technical procurement team to discuss how this optimized route can benefit your specific supply chain needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic advantages of switching to this methodology. We encourage potential partners to contact us for specific COA data and route feasibility assessments to validate the performance of this process against your current standards.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →