Advanced Synthesis of Seven-Membered Indoloazepine CYP1B1 Inhibitors for Commercial Scale-Up
The pharmaceutical industry is constantly seeking robust and scalable pathways to access novel heterocyclic scaffolds with high therapeutic potential. Patent CN115073473B discloses a groundbreaking methodology for the synthesis of seven-membered ring indoloazepine compounds, which serve as potent inhibitors of the CYP1B1 enzyme. This enzyme is a critical target in oncology and hypertension research due to its role in metabolizing pro-carcinogens and regulating vascular tone. The disclosed technology offers a significant leap forward by constructing these complex heterocycles through an efficient palladium-catalyzed C-H functionalization strategy. Unlike previous iterations that relied on restrictive substrate requirements, this invention enables the use of widely available starting materials without the need for specific ortho-substituents to drive the reaction. The resulting compounds exhibit exceptional bioactivity, with lead candidates demonstrating IC50 values as low as 10.4 nM, positioning them as prime candidates for drug development pipelines.
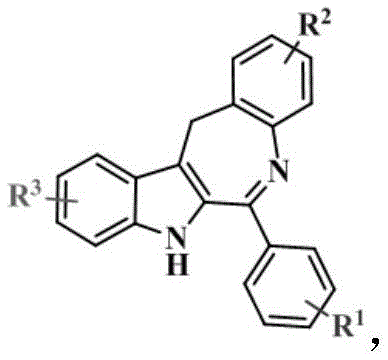
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of nitrogen-containing heterocycles via transition metal catalysis has faced significant hurdles regarding substrate scope and reaction conditions. Prior art, such as the work by Zhu's group in 2015, demonstrated palladium-catalyzed functionalization of isocyanides to build phenanthridine systems. However, extending this logic to seven-membered rings often required substrates with specific steric or electronic properties to facilitate the cyclization step. Specifically, conventional approaches frequently necessitated the presence of methyl or chlorine substituents at the ortho-position of the isocyanide group to promote the C-H activation and subsequent ring closure. This requirement drastically limits the diversity of the chemical library that can be generated, as synthesizing these ortho-substituted precursors often involves additional synthetic steps, increasing both cost and waste. Furthermore, older methodologies often relied on harsh reaction environments, utilizing strong acids, strong bases, or high-boiling polar aprotic solvent mixtures like DMF and DMSO, which complicate downstream purification and pose environmental and safety challenges in a manufacturing setting.
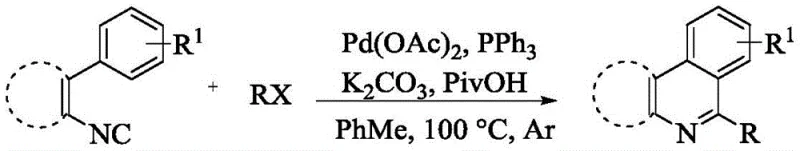
The Novel Approach
The methodology outlined in the patent data represents a paradigm shift in heterocycle synthesis by eliminating the dependency on ortho-substituted isocyanides. This new approach leverages a refined palladium catalytic system that activates the C-H bond directly without the need for steric assistance from neighboring groups. By removing this structural constraint, the process allows for a much broader range of R1, R2, and R3 substituents, including hydrogen, alkyl, alkoxy, halogens, and complex aromatic systems like pyrene or quinoline. The reaction conditions are notably milder, operating effectively in solvents such as trifluorotoluene, acetonitrile, or toluene at temperatures ranging from 90 to 150 °C under an inert argon atmosphere. This shift not only simplifies the supply chain for raw materials but also enhances the overall atom economy of the synthesis. The ability to generate diverse analogs from a common intermediate streamlines the structure-activity relationship (SAR) studies essential for optimizing drug candidates, providing medicinal chemists with a powerful tool to explore chemical space efficiently.
Mechanistic Insights into Palladium-Catalyzed C(sp2)-H Seven-Membered Ring Imidization
The core of this technological advancement lies in the intricate catalytic cycle that drives the formation of the seven-membered ring. The mechanism initiates with the oxidative addition of an aryl triflate to a palladium(0) species, generating a reactive palladium(II) intermediate. This species then undergoes coordination and migratory insertion with the isocyanide functionality, forming a key imino-palladium complex. From this junction, the reaction pathway diverges into two potential routes, both leading to the desired product but highlighting the robustness of the system. In the primary pathway, the indole moiety performs a nucleophilic attack on the palladium center, resulting in a dearomatized palladacycle intermediate. Subsequent reductive elimination releases the palladium(0) catalyst back into the cycle and generates a spirocyclic intermediate, which then undergoes a 1,2-rearrangement and aromatization to yield the final indoloazepine product. Alternatively, a Bischler-Napieralski type cyclization may occur via a highly active nitrogen intermediate if the palladium species dissociates, ensuring high conversion rates even under varying conditions. This dual-pathway capability ensures that the reaction remains efficient across a wide array of substrate electronic profiles.
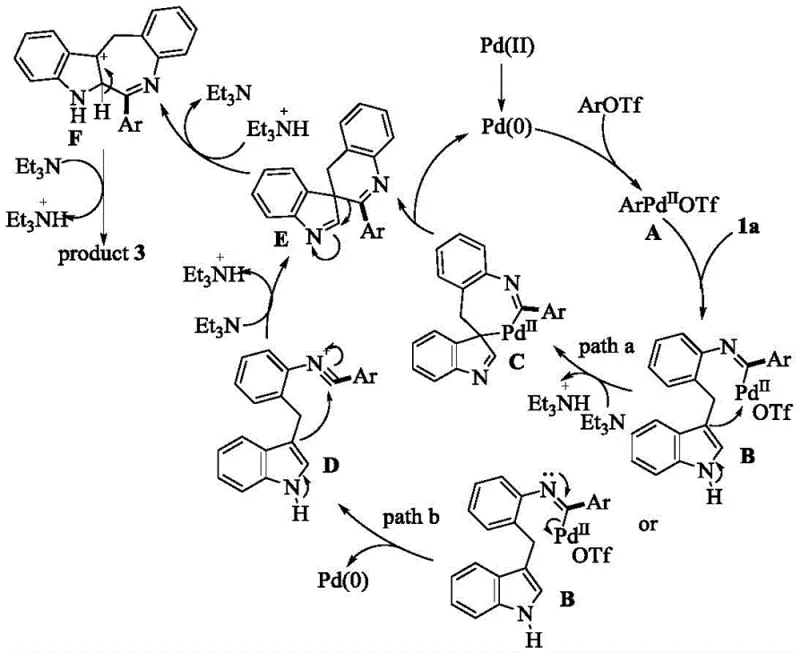
Understanding the impurity profile is critical for pharmaceutical manufacturing, and this mechanism offers inherent advantages in purity control. The specificity of the palladium insertion and the subsequent cyclization steps minimizes the formation of regioisomers that often plague C-H activation reactions. The use of aryl triflates as coupling partners, rather than halides, provides a superior leaving group ability that accelerates the oxidative addition step, reducing the residence time of reactive intermediates that could lead to side reactions. Furthermore, the mild basic conditions employed during the cyclization, typically using triethylamine, prevent the degradation of sensitive functional groups that might be present on the indole or the aryl coupling partner. This selectivity translates to a cleaner crude reaction mixture, reducing the burden on purification processes such as column chromatography or recrystallization. For process chemists, this means higher isolated yields and a more consistent quality profile, which is essential when scaling up from gram-scale discovery to kilogram-scale production for clinical trials.
How to Synthesize Indoloazepine Derivatives Efficiently
The synthesis of these high-value intermediates follows a logical five-step sequence designed for operational simplicity and high throughput. The process begins with the reduction of readily available 2-aminobenzoic acid derivatives to their corresponding alcohols, followed by a condensation with indole to establish the carbon framework. Subsequent formylation and dehydration steps convert the amine into the crucial isocyanide functionality, which serves as the handle for the final ring-closing event. The final step involves the palladium-catalyzed coupling with aryl triflates, which constructs the seven-membered azepine ring in a single operation. This convergent strategy allows for the late-stage diversification of the molecule, enabling the rapid generation of libraries for biological screening. The detailed standardized synthesis steps are provided in the guide below to ensure reproducibility and safety during implementation.
- Reduction of 2-aminobenzoic acid derivatives using LiAlH4 to form 2-aminobenzyl alcohol intermediates.
- Condensation of the alcohol with indole derivatives using trifluoroacetic acid catalyst in 1,2-dichloroethane.
- Formylation of the amine intermediate using formic acid and acetic anhydride to generate the formamide precursor.
- Dehydration of the formamide using POCl3 and triethylamine to yield the critical isocyanide intermediate.
- Final palladium-catalyzed C-H cyclization with aryl triflates to construct the seven-membered indoloazepine core.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this synthetic route offers substantial strategic benefits that translate directly to cost efficiency and supply security. The elimination of the requirement for ortho-substituted isocyanides removes a significant bottleneck in raw material sourcing. Ortho-substituted aromatics often command higher prices due to lower availability and more complex manufacturing processes compared to their unsubstituted counterparts. By utilizing generic, commodity-grade starting materials, manufacturers can significantly reduce the cost of goods sold (COGS). Additionally, the avoidance of harsh reagents like strong mineral acids or exotic mixed solvent systems simplifies the engineering controls required for production. Standard stainless steel reactors can be used without the need for specialized glass-lined equipment often necessary for handling corrosive acidic conditions, thereby lowering capital expenditure and maintenance costs associated with reactor corrosion.
- Cost Reduction in Manufacturing: The streamlined synthesis reduces the total number of unit operations and eliminates the need for expensive, specialized precursors. By avoiding the synthesis of ortho-substituted isocyanides, the process cuts out entire synthetic steps that would otherwise contribute to yield loss and waste generation. The use of common solvents like toluene and THF, which are easily recovered and recycled, further enhances the economic viability of the process. This reduction in material intensity and waste disposal costs leads to a leaner manufacturing footprint, allowing for competitive pricing in the global market for pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: Relying on broadly available starting materials mitigates the risk of supply disruptions. Specialty chemicals with narrow specifications often have limited supplier bases, creating vulnerability in the supply chain. In contrast, the precursors for this route, such as aminobenzoic acids and indoles, are produced by multiple vendors globally, ensuring a stable and continuous supply. The robustness of the reaction conditions also means that the process is less sensitive to minor variations in reagent quality, reducing the rate of batch failures and ensuring consistent delivery schedules to downstream customers.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, utilizing conditions that are safe and manageable in large-volume reactors. The absence of high-pressure hydrogenation or cryogenic steps simplifies the operational complexity. Furthermore, the improved atom economy and reduced solvent usage align with green chemistry principles, facilitating easier compliance with increasingly stringent environmental regulations. This sustainability aspect is becoming a key differentiator for suppliers, as pharmaceutical companies prioritize partners who can demonstrate a commitment to reducing the environmental impact of their supply chains.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these indoloazepine compounds. These answers are derived directly from the patented technology and experimental data to provide clarity on the feasibility and performance of the synthesis. Understanding these details helps stakeholders make informed decisions about integrating this technology into their development pipelines.
Q: What is the key advantage of this synthesis over conventional methods?
A: Unlike traditional methods requiring ortho-substituted isocyanides to promote cyclization, this novel route utilizes unsubstituted isocyanides, significantly expanding substrate scope and reducing raw material costs.
Q: What is the biological potency of the synthesized compounds?
A: The synthesized indoloazepine derivatives demonstrate high inhibitory activity against CYP1B1 enzymes, with optimal compounds achieving IC50 values as low as 10.4 nM, making them highly effective for oncology research.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the process avoids harsh conditions like strong acids or mixed high-boiling solvents, utilizing standard reagents and moderate temperatures (0-150°C), which facilitates safe and efficient commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Indoloazepine Supplier
The development of potent CYP1B1 inhibitors represents a significant opportunity in the field of oncology therapeutics, and having a manufacturing partner capable of executing complex heterocyclic synthesis is vital. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition smoothly from benchtop discovery to full-scale manufacturing. Our facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of indoloazepine intermediate meets the highest standards required for clinical and commercial applications. We understand the critical nature of timeline and quality in drug development and are committed to supporting your success.
We invite you to engage with our technical procurement team to discuss how this advanced synthesis route can optimize your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain specific insights into how switching to this methodology can reduce your overall production costs. We encourage you to contact us for specific COA data and route feasibility assessments tailored to your target molecules. Let us help you secure a reliable supply of high-quality pharmaceutical intermediates while driving down costs and accelerating your time to market.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →