Advanced Synthetic Route for High-Purity Fulvestrant Intermediates and Commercial Scalability
Advanced Synthetic Route for High-Purity Fulvestrant Intermediates and Commercial Scalability
The global pharmaceutical landscape for oncology treatments continues to evolve, driven by the urgent need for more effective therapies against hormone receptor-positive metastatic breast cancer. Fulvestrant, a potent Estrogen Receptor (ER) down-regulator, stands as a cornerstone in this therapeutic area, yet its complex molecular architecture has historically posed significant challenges for synthetic chemists and manufacturing teams alike. Recent advancements detailed in patent CN110938107B introduce a transformative approach to synthesizing fulvestrant intermediates, leveraging a robust alkylation strategy that bypasses the limitations of earlier methodologies. This technical breakthrough not only enhances the chemical purity of the final active pharmaceutical ingredient (API) but also streamlines the production workflow, offering a compelling value proposition for stakeholders across the drug development spectrum. By utilizing estradiol as a starting raw material and employing specific protective group strategies, the disclosed process achieves high yields under remarkably mild conditions.
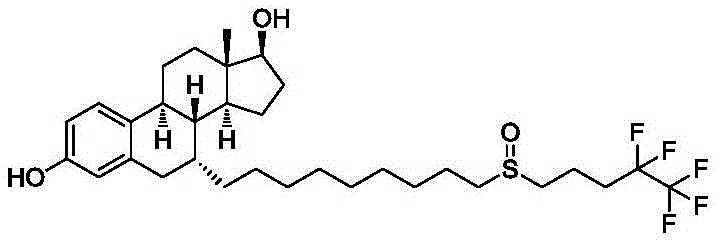
The significance of this innovation extends beyond mere academic interest; it addresses critical bottlenecks in the supply chain of high-purity pharmaceutical intermediates. As demand for endocrine therapies grows, particularly in markets with increasing incidence rates of breast cancer, the ability to produce fulvestrant reliably and cost-effectively becomes a strategic imperative. The patent outlines a sequence that moves from protected estradiol derivatives through oxidative and condensation steps, culminating in a final deprotection and oxidation to yield the target molecule. This comprehensive pathway ensures that every stereocenter is meticulously controlled, resulting in a product that meets stringent regulatory specifications for isomeric purity and chemical identity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of fulvestrant has been plagued by inefficient routes that rely on hazardous reagents and cumbersome purification protocols. For instance, early methods described in WO200232922 utilized dienones as starting materials subjected to Grignard reactions. While chemically feasible, these routes suffer from harsh reaction conditions that compromise safety and scalability. Furthermore, the condensation products obtained via Grignard addition often exhibit poor stereocontrol, generating high ratios of 7-alpha to 7-beta isomers (approximately 1.9:1 to 2.5:1). Correcting this imbalance necessitates up to four successive crystallization steps, leading to substantial material loss and inflated production costs. Other approaches, such as those found in WO2014064712, employ triethylboron as a catalyst. This reagent is not only highly flammable and toxic, posing severe risks to personnel safety, but also requires high-temperature reflux conditions that drive up energy consumption. Additionally, these legacy processes often yield intermediates with insufficient purity, forcing manufacturers to invest heavily in downstream processing to remove sulfone-type impurities and other byproducts.
The Novel Approach
In stark contrast, the methodology presented in patent CN110938107B offers a streamlined alternative that prioritizes safety, efficiency, and selectivity. The core innovation lies in the condensation reaction between a specific oxidized intermediate (Intermediate 3) and 1,9-diiodononane. This alkylation step is conducted in common organic solvents like toluene or tetrahydrofuran, using potassium tert-butoxide as a base. Crucially, the reaction proceeds efficiently at ambient temperatures ranging from 20-35°C, eliminating the need for energy-intensive heating or cryogenic cooling. The use of 1,9-diiodononane provides a stable and reactive side-chain precursor that couples cleanly with the steroid nucleus. This approach drastically reduces the formation of unwanted isomers, keeping the 7-beta isomer content below 5% in the intermediate stage and ultimately below 0.1% in the final API. By avoiding dangerous catalysts and minimizing purification cycles, this novel route represents a paradigm shift towards greener and more economical pharmaceutical manufacturing.
Mechanistic Insights into Base-Mediated Alkylation
The success of this synthetic route hinges on the precise execution of the nucleophilic substitution reaction that attaches the long alkyl side chain to the steroid core. Mechanistically, the process involves the deprotonation of the C7 position of the estradiol derivative by a strong non-nucleophilic base, typically potassium tert-butoxide. This generates a reactive enolate or carbanion species which then attacks the terminal carbon of the 1,9-diiodononane. The choice of base and solvent is critical; potassium tert-butoxide in toluene provides the optimal balance of basicity and solubility to drive the reaction forward without promoting elimination side reactions. The stoichiometry is carefully managed, with the diiodoalkane used in slight excess (1.5 to 2.5 equivalents) to ensure complete consumption of the valuable steroid intermediate. This kinetic control is essential for maximizing yield and minimizing the presence of unreacted starting materials that could complicate subsequent steps.
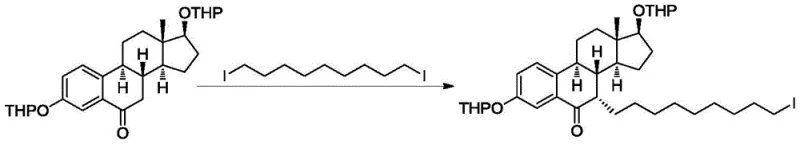
Impurity control is another pillar of this mechanism. The patent highlights the formation of sulfone-type impurities as a potential risk during the later oxidation stages. However, by ensuring high purity at the alkylation stage (Intermediate 5 or Formula I), the burden on the final purification steps is significantly reduced. The process demonstrates exceptional stereochemical fidelity, largely due to the steric environment of the steroid ring system which favors the formation of the desired 7-alpha configuration. The residual amount of Intermediate 3 in the reaction mixture is reported to be less than 3% after 8 hours, indicating a high conversion rate. This level of control is vital for a reliable pharmaceutical intermediates supplier, as it guarantees batch-to-batch consistency and simplifies the validation process for regulatory filings. The robustness of this chemistry allows for scalable production without the fear of runaway exotherms or unpredictable byproduct formation.
How to Synthesize Fulvestrant Intermediates Efficiently
Implementing this synthesis in a commercial setting requires adherence to specific operational parameters to replicate the high yields and purity reported in the patent literature. The process begins with the preparation of the reaction vessel under inert atmosphere, followed by the dissolution of the protected steroid intermediate in dry toluene. The addition of the base must be controlled to manage any initial exotherm, although the overall reaction profile is mild. Following the alkylation, the workup procedure is straightforward, involving aqueous quenching and standard liquid-liquid extraction techniques. The crude product, often obtained as an oil, is then subjected to column chromatography using mixed solvent systems like petroleum ether and ethyl acetate to isolate the pure intermediate. This purified material serves as the direct precursor for the final deprotection and oxidation steps that yield fulvestrant. For detailed operational specifics regarding reagent grades, mixing speeds, and filtration protocols, please refer to the standardized guide below.
- Dissolve the protected estradiol derivative (Intermediate 3) in an organic solvent such as toluene or tetrahydrofuran and add a strong base like potassium tert-butoxide.
- Introduce 1,9-diiodononane to the reaction mixture and maintain the temperature between 20-35°C for 8 to 10 hours to ensure complete conversion.
- Quench the reaction with aqueous ammonium chloride, perform extraction with ethyl acetate, and purify the resulting oil via silica gel column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this novel synthetic route offers tangible benefits that extend well beyond the laboratory bench. The primary advantage lies in the drastic simplification of the manufacturing process, which directly correlates to reduced operational expenditures. By eliminating the need for hazardous reagents like triethylboron and avoiding high-temperature reflux conditions, facilities can lower their insurance premiums and safety compliance costs. Furthermore, the room temperature operation significantly cuts down on energy consumption, contributing to a smaller carbon footprint and aligning with modern sustainability goals. The high conversion rates and minimal byproduct formation mean that less raw material is wasted, optimizing the cost of goods sold (COGS) for the final API. This efficiency makes the process highly attractive for cost reduction in pharmaceutical intermediates manufacturing, allowing companies to remain competitive in a price-sensitive market.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the reduction in purification steps lead to substantial cost savings. Traditional routes often require multiple crystallizations to achieve acceptable purity, each step incurring losses in yield and increases in solvent usage. In contrast, this new method achieves high purity with a single chromatographic purification, significantly lowering solvent recovery costs and waste disposal fees. The use of commodity chemicals like 1,9-diiodononane and potassium tert-butoxide ensures that raw material costs remain stable and predictable, shielding the supply chain from volatility associated with specialty reagents.
- Enhanced Supply Chain Reliability: Stability is a key concern in the sourcing of complex intermediates. Previous methods relied on side-chain iodides that were thermally unstable and difficult to store, creating bottlenecks in production scheduling. The reagents used in this patent are commercially available and stable, ensuring a continuous flow of materials. The robustness of the reaction conditions means that production can proceed without the risk of batch failures due to sensitive temperature excursions. This reliability is crucial for maintaining commercial scale-up of complex pharmaceutical intermediates, ensuring that clinical and commercial demands are met without interruption.
- Scalability and Environmental Compliance: Scaling a chemical process often introduces new challenges, but the mild nature of this alkylation reaction facilitates easy transfer from pilot plant to full-scale production. The absence of toxic heavy metals simplifies the environmental impact assessment and reduces the burden on wastewater treatment facilities. Moreover, the high selectivity of the reaction minimizes the generation of hazardous waste streams. This alignment with green chemistry principles not only satisfies regulatory requirements but also enhances the corporate social responsibility profile of the manufacturing entity, making it a preferred partner for global pharmaceutical companies seeking sustainable supply chains.
Frequently Asked Questions (FAQ)
Understanding the technical nuances of this synthesis is essential for stakeholders evaluating its adoption. The following questions address common inquiries regarding the process parameters, quality control, and scalability. These answers are derived directly from the experimental data and claims within the patent documentation, providing a factual basis for decision-making. Whether you are concerned about the specific isomeric ratios or the feasibility of large-batch production, the insights below clarify how this technology overcomes historical hurdles in fulvestrant synthesis.
Q: What are the critical temperature controls for the alkylation step in this fulvestrant process?
A: According to patent CN110938107B, the reaction is optimally conducted at mild temperatures between 20-35°C. This low-temperature regime is crucial for minimizing side reactions and controlling the formation of the 7-beta isomer, ensuring high stereochemical purity without the need for excessive energy input.
Q: How does this method improve impurity profiles compared to traditional Grignard routes?
A: Traditional methods often struggle with high isomer ratios requiring multiple crystallizations. This novel alkylation approach strictly controls the 7-beta isomer content to less than 5% in the intermediate and less than 0.1% in the final product, significantly simplifying downstream purification and reducing material waste.
Q: Is the 1,9-diiodononane reagent stable for large-scale manufacturing?
A: Yes, unlike some prior art side-chain iodines which are thermally unstable and difficult to purify, 1,9-diiodononane is a robust reagent. Its stability facilitates safer handling and consistent reaction kinetics, making the process highly suitable for industrial mass production and reliable supply chain management.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Fulvestrant Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic technologies to meet the evolving needs of the global healthcare industry. Our team of expert chemists has thoroughly analyzed the pathway described in CN110938107B and possesses the technical capability to implement this efficient route at scale. We understand that bringing a life-saving oncology drug to market requires a partner who can guarantee both quality and quantity. Therefore, we leverage our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production to ensure that your supply needs are met with precision. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, including the critical control of 7-beta isomers and sulfone impurities to levels below 0.1%.
We invite you to collaborate with us to optimize your supply chain for fulvestrant and related intermediates. By partnering with NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. Our technical procurement team is ready to provide specific COA data and route feasibility assessments to demonstrate how this innovative process can enhance your project's bottom line. Contact us today to discuss how we can support your journey from clinical development to commercial launch with reliable, high-quality pharmaceutical intermediates.