Advanced Synthesis of Aminopeptidase Inhibitors for Antitumor Drug Development
The pharmaceutical industry's relentless pursuit of effective antitumor agents has placed aminopeptidase N (APN), also known as CD13, at the forefront of oncology research. As a zinc-dependent metalloprotease highly expressed on the surface of tumor cells, APN plays a critical role in tumor invasion and metastasis by degrading the extracellular basement membrane. Consequently, the development of potent small-molecule inhibitors targeting this enzyme represents a strategic imperative for modern drug discovery. Patent CN101284820A discloses a novel class of aminopeptidase inhibitors characterized by a unique peptidomimetic structure featuring a 1,3,4-thiadiazole core linked to a chiral amino acid moiety. This technological breakthrough offers a robust platform for generating diverse analogs through systematic variation of substituents on the aromatic rings, as illustrated in the general chemical formula below.
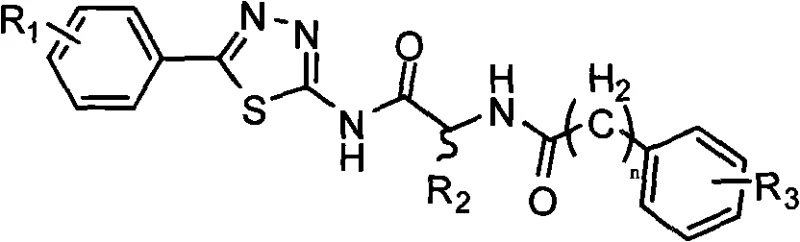
For R&D directors and procurement specialists seeking a reliable aminopeptidase inhibitor supplier, understanding the underlying synthetic methodology is paramount. The disclosed compounds are designed as small molecular analogue peptide compounds, engineered to mimic the transition state of the natural substrate while offering superior metabolic stability. The patent outlines a concise synthetic strategy that bypasses the complexities often associated with traditional peptide synthesis, such as extensive protecting group manipulations. By leveraging a convergent approach that couples a heterocyclic amine with an activated chiral acid, this method achieves a balance between structural complexity and manufacturing feasibility. This report provides a deep technical analysis of this synthesis, highlighting its potential for cost reduction in pharmaceutical intermediate manufacturing and its scalability for commercial production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional approaches to synthesizing peptidomimetic inhibitors often suffer from significant inefficiencies that hinder their translation from benchtop discovery to commercial scale. Conventional peptide coupling frequently requires the use of expensive coupling reagents and rigorous protection-deprotection sequences to prevent side reactions and maintain stereochemical purity. These multi-step processes not only extend the overall lead time but also result in cumulative yield losses that drastically increase the cost of goods sold (COGS). Furthermore, many legacy routes rely on hazardous solvents or difficult-to-remove heavy metal catalysts, creating substantial environmental compliance burdens and necessitating complex downstream purification protocols. For supply chain managers, these factors translate into volatile pricing and potential disruptions in the availability of high-purity pharmaceutical intermediates. The reliance on fragile intermediates that require cryogenic storage or immediate use further complicates logistics, making it challenging to establish a resilient supply chain for clinical trial materials.
The Novel Approach
In stark contrast, the methodology presented in CN101284820A introduces a streamlined synthetic route that addresses these historical bottlenecks through intelligent molecular design and process optimization. The core innovation lies in the construction of the 1,3,4-thiadiazole scaffold directly from commodity chemicals like p-toluic acid and thiosemicarbazide, eliminating the need for pre-functionalized heterocyclic building blocks. This approach significantly shortens the synthetic sequence, reducing the number of unit operations and minimizing solvent consumption. The subsequent coupling step utilizes a standard carbodiimide-mediated activation strategy (DCC/NHS) which is well-understood and easily scalable in industrial reactors. By avoiding exotic reagents and focusing on robust, high-yielding transformations, this novel approach facilitates the commercial scale-up of complex peptidomimetics. The ability to introduce diversity at the R1, R2, and R3 positions allows for rapid Structure-Activity Relationship (SAR) exploration without necessitating a complete overhaul of the synthetic process, thereby accelerating the drug discovery timeline.
Mechanistic Insights into Thiadiazole Formation and Amide Coupling
The synthesis begins with the formation of the key heterocyclic intermediate, 5-p-tolyl-2-amino-1,3,4-thiadiazole. This transformation is driven by the reaction of p-toluic acid with thiosemicarbazide in the presence of phosphorus oxychloride (POCl3). Mechanistically, POCl3 acts as a powerful dehydrating agent and chlorinating reagent, facilitating the cyclodehydration of the acyl thiosemicarbazide intermediate. The reaction is typically conducted under reflux conditions, where the thermal energy overcomes the activation barrier for ring closure. Following the initial cyclization, hydrolysis with water converts the intermediate chloro-species into the desired amino-thiadiazole. This step is critical as it generates the nucleophilic amine required for the subsequent coupling reaction. The use of POCl3, while requiring careful handling, ensures a high conversion rate and produces a crystalline product that can be easily purified by recrystallization from ethanol, effectively removing organic impurities and unreacted starting materials.
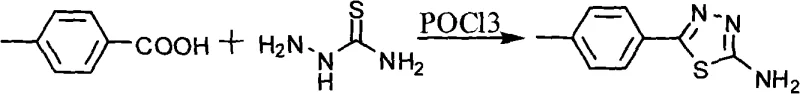
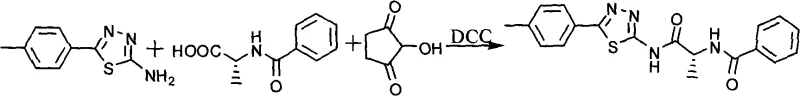
The second phase of the synthesis involves the preparation of the chiral acyl donor, benzoyl-D-alanine. This is achieved by activating benzoic acid with N-hydroxysuccinimide (NHS) and dicyclohexylcarbodiimide (DCC) to form an active ester in situ. This active ester is then reacted with D-alanine in a biphasic or aqueous-organic system buffered with sodium bicarbonate. The choice of D-alanine is strategic, as the stereochemistry at the alpha-carbon is crucial for binding affinity to the APN active site. The mild basic conditions (pH adjustment with NaHCO3) ensure that the amino group of the alanine is sufficiently nucleophilic to attack the activated ester without causing racemization. The resulting benzoyl-D-alanine precipitates upon acidification, allowing for isolation via simple filtration. This solid-phase isolation is advantageous for large-scale processing as it avoids energy-intensive distillation steps.
How to Synthesize Aminopeptidase Inhibitor Efficiently
The final assembly of the target molecule involves the coupling of the two key fragments: the 5-p-tolyl-2-amino-1,3,4-thiadiazole and the activated benzoyl-D-alanine derivative. In the preferred embodiment, the carboxylic acid of the benzoyl-D-alanine is re-activated using DCC and NHS in dioxane to generate a highly reactive succinimidyl ester. This activated species is then treated with the thiadiazole amine at room temperature. The reaction proceeds through a nucleophilic acyl substitution mechanism, forming the stable amide bond that links the heterocyclic head group to the peptidic tail. The use of dioxane as a solvent ensures good solubility for both organic fragments, promoting homogeneous reaction kinetics. Upon completion, the reaction mixture is worked up by washing with saturated sodium carbonate and dilute acid to remove urea byproducts (dicyclohexylurea) and unreacted acids. The final product is isolated by precipitation and recrystallized from a methanol-acetonitrile mixture to achieve pharmaceutical-grade purity.
- Synthesize 5-p-tolyl-2-amino-1,3,4-thiadiazole by reacting p-toluic acid with thiosemicarbazide using phosphorus oxychloride (POCl3) under reflux conditions.
- Prepare benzoyl-D-alanine by activating benzoic acid with N-hydroxysuccinimide (NHS) and DCC, followed by coupling with D-alanine in aqueous bicarbonate solution.
- Couple the activated benzoyl-D-alanine with the thiadiazole amine using DCC/NHS mediation in dioxane to form the final peptidomimetic inhibitor.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition from laboratory curiosity to commercial viability hinges on economic feasibility and operational reliability. The synthetic route described in this patent offers distinct advantages that align with the goals of cost reduction in pharmaceutical intermediate manufacturing. By utilizing commodity feedstocks such as benzoic acid, p-toluic acid, and alanine, the process decouples production costs from the volatility of specialized fine chemical markets. The elimination of complex protecting group strategies not only reduces the quantity of reagents required but also minimizes waste generation, leading to a greener and more cost-efficient process profile. Furthermore, the reliance on standard unit operations—reflux, filtration, and recrystallization—means that the process can be executed in existing multipurpose chemical plants without the need for capital-intensive equipment upgrades.
- Cost Reduction in Manufacturing: The economic benefits of this route are derived primarily from the high atom economy of the thiadiazole formation and the avoidance of expensive chiral catalysts. Since the chirality is introduced via the purchase of D-alanine, a widely available bulk chemical, the need for asymmetric synthesis or resolution steps is obviated. This simplification drastically lowers the technical barrier to entry and reduces the overall processing time. Additionally, the ability to isolate intermediates as solids allows for flexible batch scheduling and inventory management, enabling manufacturers to optimize production runs based on demand rather than the stability constraints of liquid intermediates.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the use of non-proprietary, off-the-shelf raw materials. Unlike routes that depend on custom-synthesized building blocks with long lead times, this method relies on chemicals that are produced globally at multi-ton scales. This abundance ensures a continuous supply stream and mitigates the risk of single-source dependency. The robustness of the chemistry also implies a lower failure rate during production, reducing the likelihood of batch rejections that can disrupt downstream drug formulation timelines. For global buyers, this translates to a more predictable delivery schedule and reduced exposure to supply shocks.
- Scalability and Environmental Compliance: From an environmental and safety perspective, the process is designed for scalability. The solvents employed, such as dioxane, ethanol, and ethyl acetate, are common industrial solvents with established recovery and recycling protocols. The absence of heavy metal catalysts simplifies the purification of the final API, ensuring that residual metal levels remain well below regulatory thresholds without the need for specialized scavenging resins. The solid-state nature of the intermediates and final product facilitates safe handling and transport, reducing the logistical hazards associated with shipping hazardous liquids. This alignment with green chemistry principles supports corporate sustainability goals and eases the regulatory approval process for new drug applications.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these aminopeptidase inhibitors. These insights are derived directly from the experimental data and process descriptions found in the patent literature, providing a factual basis for decision-making.
Q: What is the primary advantage of this synthetic route for aminopeptidase inhibitors?
A: The primary advantage lies in the short synthetic pathway and the use of readily available, low-cost raw materials such as p-toluic acid and thiosemicarbazide, which significantly simplifies the manufacturing process compared to traditional peptide synthesis methods.
Q: How is stereochemical integrity maintained during the synthesis?
A: Stereochemical integrity is preserved by utilizing D-alanine as the chiral starting material and employing mild coupling conditions (DCC/NHS activation at room temperature) that minimize the risk of racemization during the amide bond formation.
Q: What are the key purification methods used to ensure high purity?
A: The process utilizes multiple recrystallization steps, including ethanol recrystallization for the thiadiazole intermediate and methanol-acetonitrile recrystallization for the final product, ensuring the removal of unreacted starting materials and side products.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Aminopeptidase Inhibitor Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the successful development of antitumor therapies. Our team of process chemists has extensively analyzed the synthetic pathways disclosed in CN101284820A and possesses the expertise to optimize this route for industrial production. We offer extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can seamlessly transition from preclinical studies to late-stage clinical trials and beyond. Our state-of-the-art facilities are equipped with rigorous QC labs capable of performing comprehensive impurity profiling and stereochemical analysis, guaranteeing that every batch meets stringent purity specifications required by global regulatory agencies.
We invite you to collaborate with us to leverage this innovative chemistry for your drug discovery programs. Whether you require custom synthesis of specific analogs for SAR studies or bulk manufacturing of the lead compound, our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Contact our technical procurement team today to request specific COA data and discuss route feasibility assessments that will accelerate your path to market.