Advanced Synthesis of Aminopeptidase Inhibitors for Oncology Drug Development
The landscape of oncology drug discovery has increasingly focused on the inhibition of Aminopeptidase N (APN), also known as CD13, due to its critical role in tumor angiogenesis and metastasis. Patent CN101284820B introduces a robust and chemically elegant methodology for synthesizing a novel class of small molecule peptidomimetic compounds designed to inhibit this enzyme effectively. These compounds feature a distinctive 1,3,4-thiadiazole scaffold linked to a benzoylated amino acid moiety, offering a unique structural motif that differs significantly from traditional peptide-based inhibitors. The general chemical architecture, as depicted in the patent, allows for extensive structural diversification at the R1, R2, and R3 positions, enabling medicinal chemists to fine-tune pharmacokinetic properties and binding affinity.
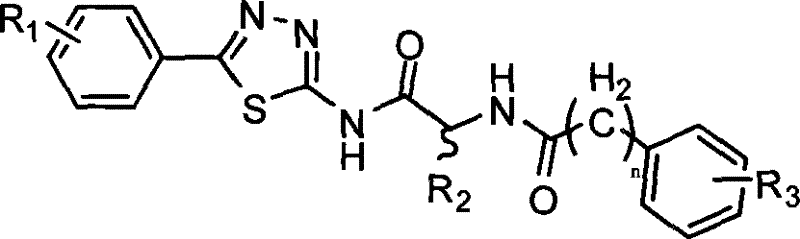
For pharmaceutical companies seeking a reliable aminopeptidase inhibitor supplier, understanding the underlying synthetic feasibility is paramount. This patent outlines a convergent strategy that bypasses the lengthy linear syntheses often associated with peptidomimetics. By breaking the molecule into two key fragments—a heterocyclic amine and an activated amino acid derivative—the process achieves high modularity. This approach not only accelerates the lead optimization phase but also lays a solid foundation for commercial scale-up of complex pharmaceutical intermediates. The ability to rapidly generate analogs with varying electronic and steric properties makes this platform technology highly valuable for developing next-generation antitumor agents.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional approaches to synthesizing APN inhibitors often rely heavily on natural amino acid derivatives coupled through standard peptide bond formation. While effective, these methods frequently suffer from low atom economy and the necessity for extensive use of protecting groups to prevent side reactions at the amine and carboxylic acid functionalities. Furthermore, many conventional routes utilize expensive coupling reagents or transition metal catalysts that require rigorous removal steps to meet stringent purity specifications for clinical candidates. The reliance on linear synthesis can also lead to cumulative yield losses, where the overall efficiency drops precipitously as the molecular complexity increases. Additionally, the solubility profiles of long peptide chains can complicate purification, often necessitating preparative HPLC which is cost-prohibitive at larger scales.
The Novel Approach
The methodology described in CN101284820B offers a transformative solution by employing a short, convergent synthetic route that minimizes these operational bottlenecks. Instead of building the molecule step-by-step from a single end, the process constructs the core thiadiazole ring independently and couples it with a pre-activated amino acid fragment. This strategy drastically reduces the number of isolation steps and eliminates the need for complex orthogonal protection schemes. The use of phosphorus oxychloride for ring closure and DCC/NHS for amide activation represents a balance between reactivity and cost-effectiveness. Consequently, this novel approach facilitates cost reduction in pharmaceutical intermediate manufacturing by streamlining the workflow and utilizing commodity chemicals that are globally accessible, thereby mitigating supply chain risks associated with specialized reagents.
Mechanistic Insights into Thiadiazole Formation and Peptide Coupling
The synthesis begins with the construction of the heterocyclic core, a critical determinant of the inhibitor's biological activity. As illustrated in the reaction scheme below, p-toluic acid undergoes a condensation reaction with thiosemicarbazide in the presence of phosphorus oxychloride (POCl3). This step is mechanistically driven by the dehydration capability of POCl3, which promotes the cyclization to form the 1,3,4-thiadiazole ring system. The reaction is typically conducted under reflux conditions, initially at 75°C followed by hydrolysis at 110°C, ensuring complete conversion to the 5-p-tolyl-2-amino-1,3,4-thiadiazole intermediate. The subsequent neutralization and recrystallization steps are vital for removing inorganic salts and unreacted starting materials, ensuring a high-purity substrate for the final coupling.
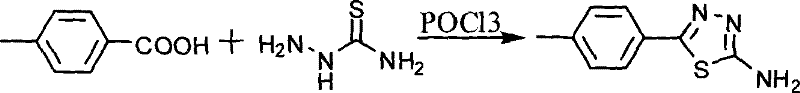
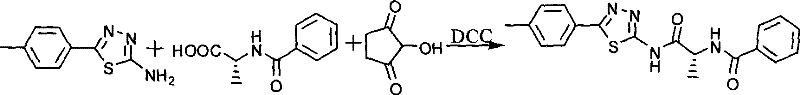
Parallel to the heterocycle synthesis, the chiral amino acid fragment is prepared to ensure the stereochemical integrity of the final product. Benzoic acid is activated using N-hydroxysuccinimide (NHS) and dicyclohexylcarbodiimide (DCC) to form an active ester intermediate. This activated species is then reacted with D-alanine under mild basic conditions to yield benzoyl-D-alanine. The use of D-alanine is crucial, as the spatial arrangement of the methyl group influences the fit within the enzyme's active site. Finally, the two fragments are united through a second DCC-mediated coupling. The active ester of benzoyl-D-alanine reacts with the amino group of the thiadiazole derivative to form the stable amide bond. This final step, shown in the following scheme, completes the assembly of the peptidomimetic scaffold.
Impurity control is inherently built into this mechanism through the physical properties of the intermediates. The patent specifies multiple recrystallization steps (using ethanol, water, or methanol-acetonitrile mixtures) which leverage the differential solubility of the product versus side products like dicyclohexylurea (DCU). This ensures that the final high-purity aminopeptidase inhibitor meets the rigorous standards required for biological testing and potential clinical application without the need for chromatographic purification.
How to Synthesize Aminopeptidase Inhibitors Efficiently
The synthesis protocol detailed in the patent provides a clear roadmap for laboratory and pilot-scale production. It emphasizes the importance of temperature control during the exothermic cyclization and the precise stoichiometry required for the coupling reactions to minimize waste. The procedure is designed to be robust, tolerating minor variations in reaction time while maintaining consistent yields. For process chemists looking to implement this route, the detailed standardized synthesis steps见下方的指南 provide the necessary operational parameters to ensure reproducibility and safety.
- Cyclize p-toluic acid with thiosemicarbazide using POCl3 to form the 5-p-tolyl-2-amino-1,3,4-thiadiazole core.
- Activate benzoic acid using N-hydroxysuccinimide (NHS) and DCC to prepare the acylating agent.
- Couple the activated benzoyl-D-alanine with the thiadiazole amine to form the final peptidomimetic inhibitor.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the value proposition of this synthetic route lies in its reliance on commodity feedstocks and simplified processing. The elimination of exotic catalysts and the use of standard organic solvents like dioxane, ethyl acetate, and methanol align perfectly with green chemistry initiatives and waste management protocols. This translates directly into operational efficiencies that benefit the bottom line without compromising on quality.
- Cost Reduction in Manufacturing: The process utilizes p-toluic acid, benzoic acid, and thiosemicarbazide, which are bulk chemicals available from multiple global suppliers at competitive prices. By avoiding precious metal catalysts or proprietary reagents, the raw material cost profile is significantly optimized. Furthermore, the high yield of the cyclization step (reported around 90% in the examples) maximizes material throughput, reducing the cost per kilogram of the final API intermediate.
- Enhanced Supply Chain Reliability: The synthetic pathway is short and convergent, meaning that the two main fragments can be manufactured in parallel, effectively halving the production lead time compared to linear sequences. The robustness of the chemistry ensures that batch-to-batch variability is minimized, providing supply chain heads with greater predictability in delivery schedules. The use of stable intermediates also allows for strategic stockpiling, further insulating the supply chain from market fluctuations.
- Scalability and Environmental Compliance: The reaction conditions operate at atmospheric pressure and moderate temperatures, removing the need for specialized high-pressure reactors. Workup procedures primarily involve filtration and crystallization, which are easily scalable unit operations in multipurpose chemical plants. The absence of heavy metals simplifies wastewater treatment and reduces the environmental footprint, facilitating easier regulatory approval for commercial manufacturing sites.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these aminopeptidase inhibitors. The answers are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and relevance for industry professionals.
Q: What is the primary advantage of this synthetic route?
A: The route utilizes readily available starting materials like p-toluic acid and avoids complex protecting group strategies, significantly simplifying the supply chain.
Q: How is stereochemistry controlled in this process?
A: Stereochemical integrity is maintained by using chiral D-alanine as the starting material for the peptide fragment, ensuring the final product retains the required optical activity.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the reaction conditions involve standard reflux temperatures and simple workup procedures like filtration and recrystallization, which are highly scalable.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Aminopeptidase Inhibitor Supplier
As the demand for targeted oncology therapies continues to surge, the need for efficient and scalable synthesis of key intermediates like aminopeptidase inhibitors has never been more critical. NINGBO INNO PHARMCHEM stands at the forefront of this sector, leveraging deep expertise in heterocyclic chemistry and peptide mimetics to deliver superior solutions. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to clinical supply is seamless. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch meets the highest international standards.
We invite you to collaborate with us to optimize your supply chain for these vital therapeutic agents. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to support your R&D and manufacturing goals, positioning your organization for success in the competitive landscape of cancer therapeutics.