Advanced Manufacturing of Ibrutinib Intermediates via Optimized Condensation and Suzuki Coupling
The pharmaceutical landscape for oncology treatments continues to evolve, with Ibrutinib standing as a cornerstone Bruton Tyrosine Kinase (BTK) inhibitor for treating Mantle Cell Lymphoma and Chronic Lymphocytic Leukemia. As demand for this critical API surges, the efficiency of its supply chain becomes paramount. Patent CN113200986A introduces a transformative preparation method for Ibrutinib and its key intermediates, addressing long-standing bottlenecks in yield and purity. This technical disclosure outlines a robust five-step synthesis starting from low-cost material flows, utilizing condensation, cyclization, substitution, Suzuki coupling, and acylation. Unlike prior art that struggles with complex purification and hazardous reagents, this innovation offers a streamlined pathway characterized by mild reaction conditions and exceptional selectivity. For R&D directors and procurement specialists, this represents a significant opportunity to optimize the commercial scale-up of complex pharmaceutical intermediates. The structural integrity of the final product, Formula G, is preserved through a sequence that rigorously controls impurity profiles.
![Chemical structure of Ibrutinib (Formula G) showing the pyrazolo[3,4-d]pyrimidine core and phenoxyphenyl moiety](/insights/img/ibrutinib-intermediate-synthesis-pharma-supplier-20260309071511-01.png)
The strategic value of this patent lies in its ability to bypass the notorious limitations of conventional synthetic routes. Historically, the synthesis of Ibrutinib has been plagued by the reliance on the Mitsunobu reaction to introduce the chiral piperidine moiety. Conventional methods, such as those reported in WO2014022390, utilize 4-aminopyrazolo[3,4-d]pyrimidine as a starting point, requiring iodination followed by Suzuki coupling and finally the problematic Mitsunobu condensation with chiral alcohols. These legacy processes are fraught with inefficiencies: the Mitsunobu step typically yields only 38%, requires stoichiometric amounts of triphenylphosphine and azodicarboxylates, and critically, demands chromatographic purification which is untenable at multi-ton scales. Furthermore, the use of tetrakis(triphenylphosphine)palladium at high loadings (0.2 equivalents) in older Suzuki steps inflates raw material costs significantly. In stark contrast, the novel approach detailed in CN113200986A initiates the synthesis with a direct condensation between Compound A and the chiral hydrazine Compound B. This fundamental shift eliminates the need for late-stage chiral introduction via Mitsunobu chemistry, thereby securing the stereochemical integrity from the outset and enabling a total yield that far exceeds the single-digit percentages of previous generations.
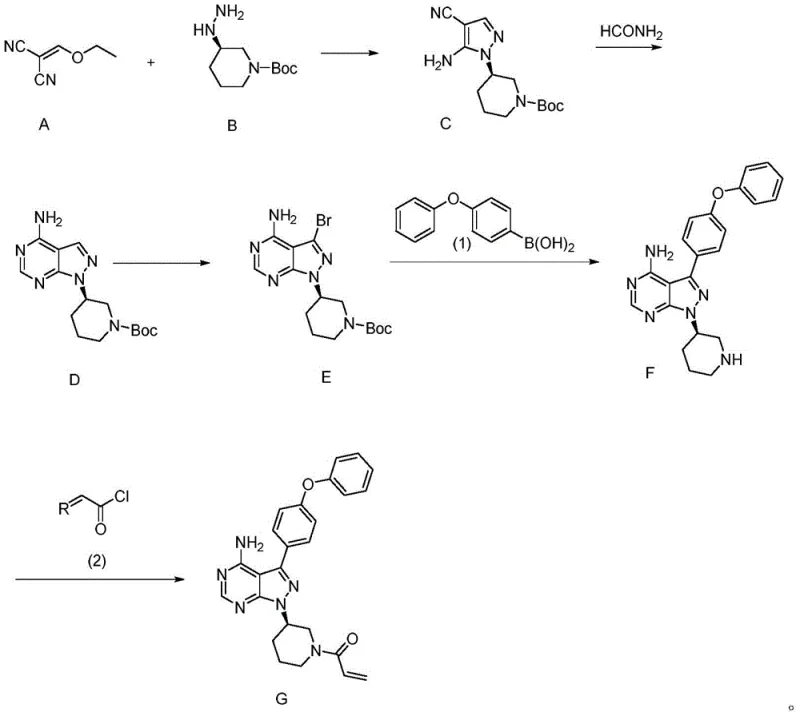
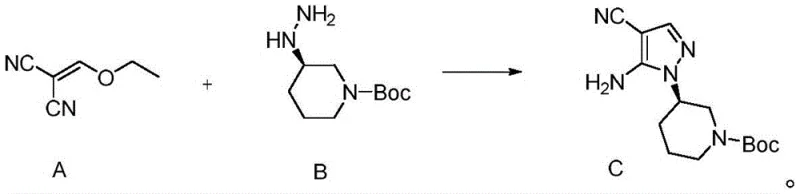
Mechanistically, the success of this route hinges on the precise control of the initial cyclization and subsequent functionalization steps. The formation of the pivotal Intermediate C is achieved through a condensation reaction between Compound A (an ethoxymethylene malononitrile derivative) and Compound B (a chiral hydrazine) in a protic solvent like ethanol. Operating at temperatures between 78°C and 85°C, this reaction proceeds with remarkable efficiency, achieving yields greater than 95% without the need for exotic catalysts. The choice of ethanol not only facilitates the dissolution of reactants but also promotes the intramolecular cyclization required to form the pyrazole ring while maintaining the chiral configuration of the piperidine nitrogen. Following this, the conversion of Compound C to Compound D involves a cyclization with formamide, effectively constructing the pyrimidine ring of the core scaffold. This step is crucial for establishing the hydrogen-bonding network necessary for BTK inhibition. Subsequent bromination of Compound D to form Compound E activates the C3 position for cross-coupling. The use of N-bromosuccinimide (NBS) or elemental bromine in DMF allows for regioselective halogenation, preparing the molecule for the critical Suzuki-Miyaura coupling.
The Suzuki coupling step, transforming Compound E into Compound F, represents another area of significant optimization. By employing a palladium catalyst loading as low as 0.01 equivalents (compared to 0.2 in prior art), the process drastically reduces heavy metal contamination risks and cost. The reaction utilizes 4-phenoxyphenylboronic acid in a biphasic system of 1,4-dioxane and water with potassium phosphate as the base. This conditions ensure high conversion rates while minimizing the formation of homocoupling byproducts. Finally, the acylation of the secondary amine in Compound F with an acryloylating agent (such as acryloyl chloride or activated esters) yields the final Ibrutinib structure. Throughout this sequence, the impurity profile is tightly managed; the avoidance of chromatographic purifications in favor of crystallizations and extractions ensures that the final API achieves a purity of over 99.5%, with single impurities controlled below 0.1%. This level of control is essential for meeting the rigorous regulatory standards required for high-purity pharmaceutical intermediates.
How to Synthesize Ibrutinib Intermediate Efficiently
Implementing this novel synthetic route requires strict adherence to the optimized reaction parameters defined in the patent to ensure reproducibility and safety. The process begins with the preparation of Compound C, where temperature control during the reflux in ethanol is critical to prevent side reactions. Following the isolation of Intermediate C, the formamide cyclization must be monitored via HPLC to ensure complete consumption of the nitrile group before proceeding to bromination. The bromination step requires careful handling of halogenating agents, with post-reaction toluene washes effectively removing unreacted bromine. For the Suzuki coupling, degassing the solvent system to remove oxygen is vital to maintain catalyst activity. Detailed standard operating procedures regarding stoichiometry, addition rates, and workup protocols are essential for transferring this chemistry from the lab to the pilot plant. The following guide outlines the standardized synthesis steps derived from the patent examples.
- Condense Compound A and Compound B in ethanol at 78-85°C to form Intermediate C with high optical purity.
- Cyclize Compound C with formamide to generate the pyrazolo[3,4-d]pyrimidine core (Compound D).
- Perform bromination on Compound D using NBS or bromine to activate the position for Suzuki coupling (Compound E).
- Execute Suzuki coupling with 4-phenoxyphenylboronic acid to install the phenoxyphenyl group (Compound F).
- Finalize the synthesis via acylation with an acryloylating agent to yield the target Ibrutinib structure.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers profound strategic advantages beyond mere technical elegance. The primary driver for cost reduction lies in the elimination of the Mitsunobu reaction, which traditionally consumes expensive and hazardous reagents like DIAD and triphenylphosphine in stoichiometric quantities. By replacing this with a direct condensation using commodity chemicals, the raw material cost per kilogram of the intermediate is significantly reduced. Furthermore, the drastic reduction in palladium catalyst loading during the Suzuki step—from 0.2 equivalents in legacy processes to merely 0.01 equivalents in this new method—translates to substantial savings in precious metal costs and simplifies the downstream removal of heavy metals, a common bottleneck in API manufacturing. The process also minimizes the generation of three wastes (waste water, gas, and solids) by avoiding chromatographic purification entirely, relying instead on crystallization and filtration which are inherently more environmentally friendly and scalable.
Supply chain reliability is markedly enhanced by the use of readily available starting materials. Compound A and Compound B are either commercially available or can be synthesized via established, high-yield routes, reducing the risk of raw material shortages. The robustness of the reaction conditions, particularly the tolerance for mild temperatures and common solvents like ethanol and toluene, ensures that the process can be executed consistently across different manufacturing sites without requiring specialized equipment. This flexibility is crucial for maintaining supply continuity in the face of global logistical disruptions. Additionally, the high yield of each step (often exceeding 90%) means that less starting material is required to produce the same amount of final product, effectively increasing the throughput of existing manufacturing facilities without the need for capital-intensive expansion. This efficiency directly supports cost reduction in pharmaceutical manufacturing by maximizing asset utilization.
Scalability and environmental compliance are seamlessly integrated into this process design. The avoidance of column chromatography is perhaps the most significant factor enabling commercial scale-up; chromatography is notoriously difficult to scale and often limits batch sizes. By designing a route where intermediates can be isolated via simple filtration or extraction, the process becomes amenable to continuous manufacturing or large-batch processing. The use of greener solvents and the reduction of hazardous waste align with increasingly stringent environmental regulations, mitigating the risk of production stoppages due to compliance issues. This makes the technology an ideal candidate for reducing lead time for high-purity API intermediates, allowing manufacturers to respond rapidly to market demand while maintaining a sustainable operational footprint.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Ibrutinib intermediate synthesis. These answers are derived directly from the experimental data and beneficial effects described in patent CN113200986A, providing clarity on process robustness and quality control. Understanding these nuances is critical for technical teams evaluating the feasibility of technology transfer.
Q: How does this new route improve upon traditional Mitsunobu reactions?
A: Traditional routes rely on Mitsunobu reactions which suffer from low yields (around 38%), require hazardous reagents like DEAD, and necessitate difficult chromatographic purification. This novel method utilizes a direct condensation strategy that achieves yields over 95%, eliminates toxic phosphine waste, and allows for simple filtration workups, drastically reducing production costs and environmental impact.
Q: What represents the critical quality attribute in the synthesis of Compound C?
A: The critical quality attribute is the maintenance of optical purity at the chiral center introduced by Compound B. The new condensation protocol operates under mild conditions (78-85°C in ethanol) that prevent racemization, ensuring the final API meets stringent enantiomeric excess specifications without the need for chiral resolution steps.
Q: Is this process suitable for multi-ton commercial scale-up?
A: Yes, the process is explicitly designed for industrial amplification. It replaces column chromatography with crystallization and filtration, uses commercially available solvents like ethanol and toluene, and employs robust catalytic systems (Pd) at low loadings, making it highly adaptable for 100 MT annual production capacities.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ibrutinib Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent literature to commercial reality requires more than just chemical knowledge; it demands engineering excellence and rigorous quality assurance. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this novel route are fully realized in practice. Our state-of-the-art facilities are equipped to handle the specific solvent systems and catalytic requirements of this synthesis, while our stringent purity specifications and rigorous QC labs guarantee that every batch of Ibrutinib intermediate meets the highest global regulatory standards. We are committed to delivering a reliable supply of this critical oncology building block, supporting your drug development and commercialization timelines with unwavering consistency.
We invite you to collaborate with us to unlock the full potential of this optimized manufacturing process. Our technical team is prepared to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how this route can improve your margins. Please contact our technical procurement team today to request specific COA data, route feasibility assessments, and sample availability. Let us be your partner in advancing affordable and accessible cancer therapies through superior chemical manufacturing.