Advanced Manufacturing of Aramchol: A High-Yield Route for Pharmaceutical Intermediates
Advanced Manufacturing of Aramchol: A High-Yield Route for Pharmaceutical Intermediates
The pharmaceutical landscape for treating Non-Alcoholic Fatty Liver Disease (NAFLD) is rapidly evolving, with Aramchol emerging as a pivotal therapeutic candidate due to its unique fatty acid-bile acid conjugate structure. As detailed in patent CN109503693B, a groundbreaking synthesis method has been developed that utilizes cholic acid and arachidic acid as primary raw materials to efficiently construct this complex molecule. This innovation addresses critical bottlenecks in previous manufacturing techniques by introducing a highly regioselective activation strategy. The core of this breakthrough lies in the precise manipulation of stereochemistry at the C-3 position of the cholic acid backbone, ensuring the final product meets the rigorous optical purity standards required for clinical applications. By leveraging steric hindrance effects through specific sulfonyl chlorides, the process achieves a level of control that was previously difficult to attain without excessive protection steps.
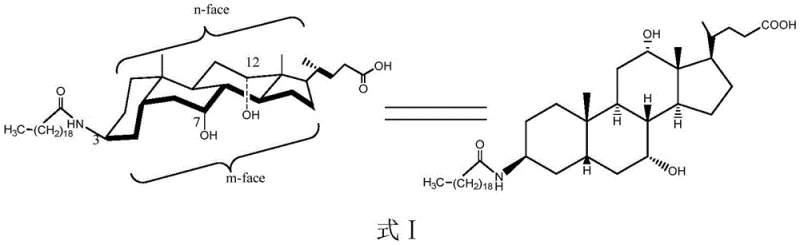
For R&D directors and process chemists, the structural integrity of the final API is paramount. The target molecule, Aramchol, is chemically defined as 3 beta-arachidoyl-7 alpha, 12 alpha-di-hydroxy-5 beta-cholestane-24-oic acid. Its efficacy is intrinsically linked to the specific spatial arrangement of its hydroxyl and amide groups. The patent highlights that maintaining the 3-beta configuration is not merely a structural preference but a biological necessity for the drug's mechanism of action in reducing liver fat content and preventing atherosclerosis. Consequently, any synthetic route must guarantee the inversion of the natural 3-alpha hydroxyl of cholic acid to the 3-beta amino configuration with absolute fidelity. This new process provides a robust framework for achieving this transformation, laying a solid foundation for the optical purity control of the final product Aramchol, which is essential for regulatory approval and patient safety.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
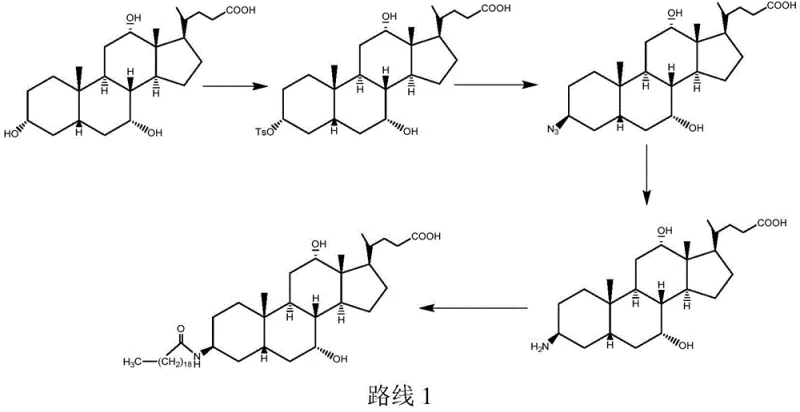
Historically, the synthesis of Aramchol has been plagued by issues related to selectivity and process length. Traditional Route 1, for instance, involves reacting cholic acid directly with p-toluenesulfonyl chloride. While conceptually simple, this approach suffers from poor reaction selectivity because the carboxyl group and other hydroxyl groups on the cholic acid molecule also react with the sulfonyl chloride. This leads to a complex mixture of side products that are notoriously difficult to separate, resulting in low yields of the desired intermediate and high impurity levels in the final drug substance. Even when strategies like carboxyl esterification are employed to mitigate some side reactions, the exposure of three active hydroxyl sites still generates significant byproduct formation. Furthermore, alternative Route 2 attempts to solve this by protecting all three hydroxyls with acetyl groups, but this introduces a cumbersome nine-step sequence. The total yield for such protected routes is often dismally low, reported around 12.7 percent based on cholic acid, making them economically unviable for large-scale manufacturing due to excessive material consumption and waste generation.
The Novel Approach
In stark contrast, the novel process disclosed in the patent offers a streamlined pathway that bypasses the need for exhaustive protection-deprotection cycles. By selecting alkyl benzene sulfonyl chlorides with specific steric properties, specifically those with bulky substituents like the tert-butyl group at the para-position, the reaction can be directed exclusively to the 3-hydroxyl group. This selectivity is driven by the subtle differences in the electronic environment and steric accessibility of the three hydroxyl groups on the cholic acid ring system. The 3-hydroxyl is slightly more active and less sterically hindered than the 7- and 12-hydroxyls. When combined with a bulky tertiary amine acid-binding agent, the reagent effectively discriminates between these sites. This strategic choice allows for the direct formation of the key 3-alpha-O-alkyl benzene sulfonyl cholic acid methyl ester intermediate in high yield. The subsequent nucleophilic substitution with sodium azide then proceeds with perfect stereochemical inversion, setting the stage for a high-efficiency synthesis that drastically reduces the number of unit operations compared to legacy methods.
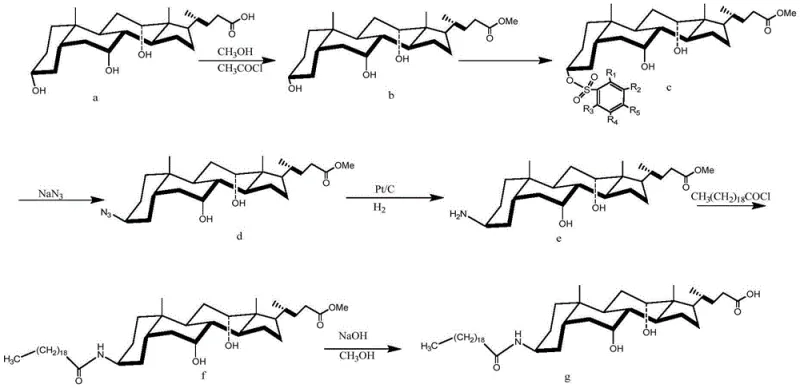
Mechanistic Insights into Regioselective Sulfonylation and Inversion
The success of this synthesis hinges on a sophisticated understanding of stereoelectronic effects during the activation and substitution phases. The selection of the activating agent is critical; the patent specifies the use of alkyl benzene sulfonyl chlorides where the para-position substituent (R5) is a bulky group such as tert-butyl or tert-amyl. This bulkiness serves a dual purpose: it prevents the reagent from accessing the more sterically crowded 7- and 12-hydroxyl positions, and it creates a leaving group that facilitates a clean SN2 reaction. During the nucleophilic substitution step with sodium azide, the azide ion acts as a strong nucleophile. Due to the steric bulk of the sulfonate leaving group and the rigid steroid backbone, the azide is forced to attack the chiral C-3 carbon from the opposite face (backside attack). This geometric constraint ensures a complete inversion of configuration from the 3-alpha sulfonate to the 3-beta azide. This mechanistic precision is what allows the process to achieve an optical purity of greater than 99 percent, eliminating the need for costly chiral chromatography or recrystallization steps that typically burden bile acid derivative synthesis.
Furthermore, the reaction conditions are optimized to support this mechanistic pathway without degrading the sensitive steroid skeleton. The use of a phase transfer catalyst, such as tetrabutylammonium bromide, in a solvent system like tert-butanol enhances the nucleophilicity of the azide ion while maintaining the solubility of the lipophilic cholic acid derivative. This homogeneous-like environment ensures that the reaction kinetics favor the desired inversion over potential elimination side reactions. The subsequent reduction of the azide to an amine using Pt/C catalytic hydrogenation is equally critical, as it must proceed without affecting the other hydroxyl groups or the newly formed stereochemistry. The careful control of pH and temperature during the final amidation and hydrolysis steps ensures that the delicate balance of functional groups is maintained, resulting in a final product that is chemically identical to the reference standard with minimal impurity profiles.
How to Synthesize Aramchol Efficiently
The practical implementation of this synthesis involves a logical sequence of transformations starting from readily available cholic acid. The process begins with the protection of the carboxylic acid as a methyl ester, followed by the crucial regioselective sulfonylation at the 3-position. Once the leaving group is installed, the stereochemical inversion is achieved via azidation, followed by reduction to the amine. The final stages involve coupling with arachidoyl chloride and deprotection of the ester. This sequence represents a significant optimization over prior art, reducing the operational complexity while maximizing yield. For process engineers looking to implement this technology, the detailed standardized synthesis steps are provided below to ensure reproducibility and quality control.
- Esterify cholic acid with methanol and acetyl chloride to protect the carboxyl group, forming methyl cholate.
- React methyl cholate with p-tert-butylbenzenesulfonyl chloride and a bulky tertiary amine to selectively activate the 3-hydroxyl group.
- Perform nucleophilic substitution with sodium azide using a phase transfer catalyst to invert the configuration at C-3, followed by catalytic hydrogenation to form the amine.
- Couple the resulting amine with arachidoyl chloride to form the amide bond, followed by alkaline hydrolysis to remove the methyl ester protection and yield Aramchol.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this novel synthesis route offers transformative benefits that directly impact the bottom line and operational resilience. The primary advantage lies in the drastic simplification of the manufacturing workflow. By eliminating the need for multiple protection and deprotection steps associated with the 7- and 12-hydroxyl groups, the process significantly reduces the consumption of reagents, solvents, and labor hours. This streamlining translates into substantial cost savings in raw material procurement and waste disposal, as fewer unit operations mean less energy consumption and lower environmental compliance costs. Additionally, the high selectivity of the reaction minimizes the formation of hard-to-remove impurities, which simplifies the purification process and increases the overall throughput of the manufacturing facility. This efficiency allows for a more competitive pricing structure for the final API intermediate, providing a distinct market advantage.
- Cost Reduction in Manufacturing: The elimination of complex protection groups means fewer reaction vessels are tied up for longer periods, and the consumption of expensive protecting reagents like acetic anhydride is removed entirely. This reduction in material intensity directly lowers the variable cost per kilogram of production. Moreover, the high yield of the key intermediates ensures that the expensive cholic acid starting material is utilized with maximum atom economy, preventing the financial loss associated with low-yielding multi-step sequences. The simplified downstream processing also reduces the load on purification equipment, extending asset life and reducing maintenance overheads.
- Enhanced Supply Chain Reliability: The reliance on robust, commodity-grade reagents such as p-tert-butylbenzenesulfonyl chloride and sodium azide ensures that the supply chain is not vulnerable to the shortages of exotic catalysts or specialized ligands. The process uses common solvents like petroleum ether and methanol, which are widely available globally, mitigating the risk of logistics disruptions. Furthermore, the high reproducibility of the reaction conditions means that batch-to-batch variability is minimized, ensuring a consistent supply of high-quality intermediate to downstream formulation partners. This reliability is crucial for maintaining uninterrupted production schedules for the final drug product.
- Scalability and Environmental Compliance: The process is inherently scalable, moving seamlessly from laboratory benchtop to pilot plant and full commercial production without significant re-engineering. The reduction in the number of steps inherently reduces the total volume of chemical waste generated, aligning with green chemistry principles and easing the burden on wastewater treatment facilities. The use of catalytic hydrogenation rather than stoichiometric metal reductions further reduces heavy metal waste. This environmental profile not only lowers disposal costs but also future-proofs the manufacturing site against increasingly stringent environmental regulations, ensuring long-term operational continuity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and mechanistic explanations provided in the patent documentation, offering clarity on the feasibility and benefits of adopting this route for commercial manufacturing.
Q: How does this process ensure high optical purity for Aramchol?
A: The process utilizes p-tert-butylbenzenesulfonyl chloride, which possesses significant steric hindrance. This bulkiness ensures that during the nucleophilic substitution with sodium azide, the azide group attacks the chiral C-3 center exclusively from the opposite direction of the leaving group. This strict stereoelectronic control guarantees the inversion of configuration required for the 3-beta-amino structure, resulting in an optical purity exceeding 99 percent without the need for chiral resolution.
Q: What are the primary advantages over conventional synthesis routes?
A: Conventional routes often suffer from poor regioselectivity, requiring extensive protection and deprotection steps for the 7- and 12-hydroxyl groups, which drastically lowers overall yield and increases waste. This novel method leverages the intrinsic reactivity differences of the hydroxyl groups combined with bulky activating agents to achieve selective reaction at the 3-position. This eliminates multiple protection steps, significantly shortening the synthetic timeline and improving the total yield compared to traditional multi-step pathways.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the route is designed with scalability in mind. It avoids the use of exotic or highly unstable reagents, relying instead on robust chemistry such as esterification, sulfonylation, and standard catalytic hydrogenation. The reaction conditions are mild and controllable, utilizing common solvents like petroleum ether and tert-butanol. Furthermore, the high selectivity reduces the formation of difficult-to-separate impurities, simplifying downstream purification and making the process economically viable for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Aramchol Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthesis routes for next-generation therapeutics like Aramchol. Our team of expert process chemists has thoroughly analyzed this patent technology and is fully equipped to translate these laboratory findings into robust commercial manufacturing processes. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. Our state-of-the-art facilities are designed to handle complex organic syntheses with stringent purity specifications, supported by rigorous QC labs that guarantee every batch meets the highest international standards. We are committed to delivering high-purity Aramchol intermediates that facilitate the rapid development and market entry of this vital NAFLD treatment.
We invite you to collaborate with us to leverage this advanced synthesis technology for your supply chain. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this streamlined route can optimize your budget. Please contact us today to request specific COA data and route feasibility assessments. Let us partner with you to secure a reliable, cost-effective, and high-quality supply of Aramchol, driving your pharmaceutical projects forward with confidence and speed.