Scalable Synthesis of Galanthamine Intermediates via Novel Benzaldehyde Derivatives
The pharmaceutical industry continuously seeks robust and scalable pathways for the production of high-value active pharmaceutical ingredients (APIs), particularly for neurodegenerative treatments where demand is rising. Patent CN102304031A introduces a significant advancement in the total synthesis of (-)-Galanthamine, a potent acetylcholinesterase inhibitor used in the management of Alzheimer's disease. This intellectual property details a novel class of 2,3,4-substituted benzaldehyde derivatives that serve as pivotal intermediates, enabling a more efficient construction of the complex tetracyclic alkaloid framework. By shifting away from reliance on natural extraction from Amaryllidaceae plants, which is plagued by low yields and supply chain volatility, this chemical synthesis route offers a reliable alternative for securing the global supply of this critical medication. The methodology leverages modern cross-coupling technologies and stereoselective reductions to achieve high purity standards essential for regulatory compliance.
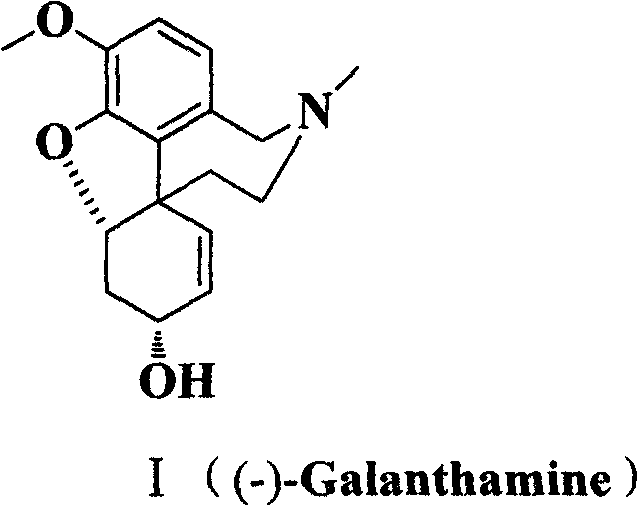
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the procurement of Galanthamine has been heavily dependent on the extraction from natural sources such as Lycoris squamigera and Lycoris aurea, a strategy that presents inherent logistical and economic vulnerabilities for pharmaceutical manufacturers. The concentration of the alkaloid within these plant matrices is exceptionally low, necessitating the processing of massive quantities of biomass to isolate therapeutic amounts, which drives up the cost of goods sold significantly. Furthermore, agricultural sourcing is subject to seasonal variations, climatic disruptions, and geographical limitations, creating an unstable supply chain that cannot guarantee the continuous availability required for commercial drug production. Existing total synthesis methods reported in prior art, while chemically interesting, often suffer from excessive step counts, the use of hazardous reagents, or conditions that are difficult to control on a multi-kilogram scale, rendering them economically unviable for industrial adoption.
The Novel Approach
The innovative strategy outlined in the patent data circumvents these bottlenecks by introducing a streamlined synthetic sequence initiated from readily available isovanillin. The core breakthrough lies in the preparation of a specific benzaldehyde derivative (Formula II) via a halogenation and vinyl ethyl ether addition sequence, which sets the stage for a highly efficient Suzuki coupling reaction. This approach constructs the biaryl bond early in the synthesis, establishing the carbon skeleton with high fidelity before proceeding to ring-closing and functional group manipulations. The entire pathway is designed with scalability in mind, utilizing relatively mild reaction temperatures and common organic solvents that simplify process engineering and waste management. By integrating a chiral resolution step followed by asymmetric reduction, the process ensures the delivery of the pharmacologically active (-)-enantiomer with high optical purity, meeting the stringent requirements for API intermediates.
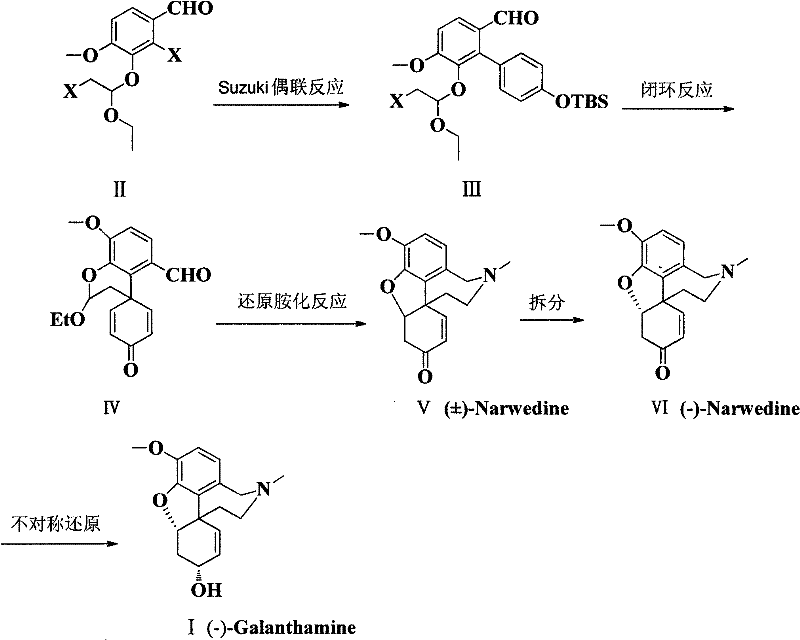
Mechanistic Insights into Suzuki Coupling and Cyclization
The chemical elegance of this route is anchored in the Suzuki-Miyaura cross-coupling reaction, which joins the novel benzaldehyde derivative with a specialized boron reagent to form the biaryl intermediate. The patent specifies the use of a tri-borane reagent protected with tert-butyldimethylsilyl (TBS) groups, which ensures compatibility with the subsequent cyclization steps. Catalyzed by palladium species such as Pd2(dba)3 in the presence of phosphine ligands like triphenylphosphine, this transformation proceeds under reflux conditions in aprotic polar solvents such as tetrahydrofuran. Following the coupling, the synthesis employs a fluoride-mediated cyclization using cesium fluoride at elevated temperatures (around 140°C) to close the ether bridge, forming the rigid tetracyclic core characteristic of the Galanthamine family. This sequence demonstrates excellent chemoselectivity, preserving the aldehyde functionality required for the downstream reductive amination while constructing the complex ring system efficiently.
Impurity control is meticulously managed through the selection of reagents and purification techniques at each stage. The reductive amination step, which converts the ketone intermediate into the amine framework of Narwedine, utilizes sodium cyanoborohydride under controlled pH conditions to minimize over-reduction or side reactions. The subsequent resolution of racemic Narwedine is achieved through a sophisticated crystallization process involving seeding with pure (-)-Narwedine crystals in a mixed solvent system of aliphatic alcohol and alkylamine. This thermodynamic control allows for the preferential precipitation of the desired enantiomer, effectively purging the unwanted (+)-isomer from the solid phase. Finally, the asymmetric reduction using L-Selectride at cryogenic temperatures (-78°C) ensures the stereochemical integrity of the final hydroxyl group, delivering the target molecule with the specific optical rotation required for biological activity.
How to Synthesize (-)-Galanthamine Efficiently
Executing this synthesis requires precise adherence to the reaction parameters defined in the patent to ensure optimal yield and purity profiles suitable for pharmaceutical applications. The process begins with the careful preparation of the halogenated benzaldehyde precursor, followed by the critical Suzuki coupling which demands anhydrous conditions and inert atmosphere handling to maintain catalyst activity. Operators must monitor the cyclization step closely to ensure complete ring closure without degrading the sensitive aldehyde moiety. The downstream processing involves standard unit operations such as extraction, drying, and column chromatography, which are well-suited for pilot and commercial plant environments. For a detailed breakdown of the specific operational parameters, stoichiometry, and workup procedures, please refer to the standardized synthesis guide below.
- Preparation of the key 2,3,4-substituted benzaldehyde derivative (Formula II) via halogenation and vinyl ethyl ether addition.
- Execution of Suzuki coupling followed by fluoride-mediated cyclization to form the tetracyclic ketone framework.
- Reductive amination to generate racemic Narwedine, followed by chiral resolution and asymmetric reduction to yield (-)-Galanthamine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route represents a strategic shift towards greater supply security and cost predictability in the manufacturing of neurological therapeutics. By replacing the variable and limited supply of plant-derived raw materials with a fully synthetic pathway based on commodity chemicals like isovanillin, manufacturers can decouple their production schedules from agricultural harvest cycles. This transition significantly mitigates the risk of raw material shortages and price volatility, ensuring a steady flow of intermediates necessary for continuous API production. Furthermore, the streamlined nature of the synthesis reduces the overall number of processing steps compared to traditional methods, which inherently lowers the consumption of solvents, energy, and labor hours per kilogram of finished product.
- Cost Reduction in Manufacturing: The elimination of expensive and inefficient extraction processes leads to substantial cost savings in the overall production budget. By utilizing widely available starting materials and avoiding the need for complex chiral pool precursors, the direct material costs are significantly optimized. Additionally, the use of robust catalytic systems and standard purification techniques reduces the need for specialized equipment or exotic reagents, further driving down the capital and operational expenditures associated with the manufacturing process.
- Enhanced Supply Chain Reliability: Relying on a chemical synthesis route ensures a consistent and predictable lead time for the delivery of high-purity pharmaceutical intermediates. Unlike botanical sources which are subject to geopolitical and environmental risks, the supply of synthetic precursors is stable and can be sourced from multiple qualified vendors globally. This redundancy strengthens the supply chain resilience, allowing pharmaceutical companies to maintain safety stock levels with confidence and meet market demand without interruption.
- Scalability and Environmental Compliance: The reaction conditions described, such as the use of THF and DMF at manageable temperatures, are highly amenable to scale-up from laboratory benchtop to multi-ton commercial reactors. The process avoids the generation of excessive hazardous waste streams associated with older synthetic methodologies, aligning with modern green chemistry principles and environmental regulations. This facilitates easier regulatory approval and reduces the burden on waste treatment facilities, making the process sustainable for long-term industrial operation.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Galanthamine synthesis technology. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing clarity on the feasibility and benefits of the route. Understanding these details is crucial for technical teams evaluating the integration of this process into their existing manufacturing portfolios.
Q: What are the advantages of this synthetic route over plant extraction?
A: Unlike plant extraction which suffers from low content and seasonal supply constraints, this total synthesis method offers consistent quality, scalable production capacity, and independence from agricultural variables.
Q: Is the Suzuki coupling step suitable for large-scale manufacturing?
A: Yes, the patent specifies mild reaction conditions using standard palladium catalysts and phosphine ligands in refluxing aprotic solvents, which are well-established parameters for industrial scale-up.
Q: How is chirality controlled in this process?
A: Chirality is established through a two-stage process: first by crystallization-induced resolution of the racemic Narwedine using seed crystals, followed by stereoselective asymmetric reduction using L-Selectride.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Galanthamine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of having a dependable partner for the production of complex pharmaceutical intermediates like those required for Galanthamine synthesis. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from development to full-scale manufacturing. We are committed to maintaining stringent purity specifications through our rigorous QC labs, guaranteeing that every batch of API intermediate meets the highest international standards for safety and efficacy. Our facility is equipped to handle the specific reaction conditions outlined in this patent, including cryogenic reductions and moisture-sensitive coupling reactions.
We invite you to collaborate with us to optimize your supply chain and reduce your overall manufacturing costs through our advanced chemical capabilities. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our expertise can support your commercial goals and ensure the uninterrupted supply of high-quality Galanthamine intermediates for your global markets.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →