Advanced Synthetic Route for Entecavir Intermediates: Enhancing Purity and Scalability for Global API Manufacturing
The pharmaceutical industry continuously seeks robust synthetic pathways for potent antiviral agents, and the synthesis of Entecavir, a carbocyclic guanosine analogue with significant activity against Hepatitis B virus, remains a critical focus. Patent CN101838207B introduces a transformative methodology that fundamentally alters the construction of the nucleoside skeleton, moving away from the historically problematic direct ring-opening of guanine derivatives. This technical insight report analyzes the strategic advantages of this novel route, which prioritizes the early formation of the carbocyclic amine intermediate before constructing the purine ring system. By decoupling the sugar moiety synthesis from the nucleobase coupling, the process achieves superior regioselectivity and simplifies downstream purification, addressing long-standing bottlenecks in the manufacturing of high-purity nucleoside analogues.
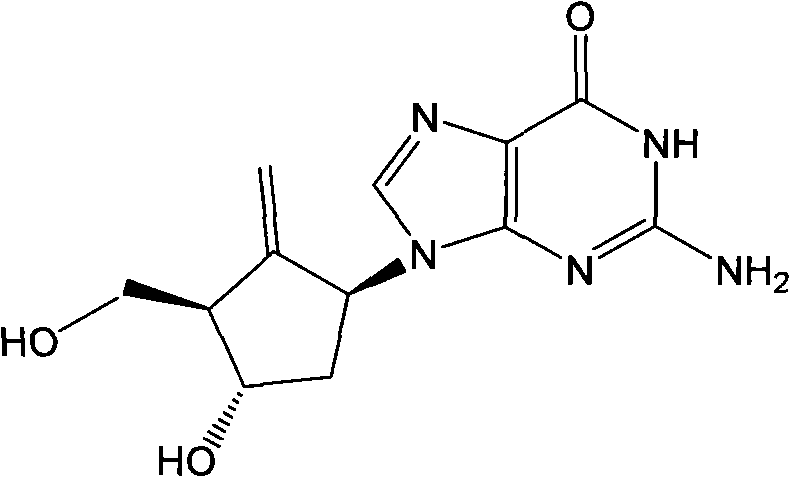
For R&D directors and process chemists, understanding the limitations of conventional methods is essential to appreciating the value of this innovation. Historically, the synthesis of carbocyclic nucleosides like Entecavir relied heavily on the direct nucleophilic attack of guanine derivatives on epoxy cyclopentane compounds. While conceptually straightforward, this approach is plagued by inherent chemical inefficiencies. The ring-opening reaction typically yields a mixture of regioisomers (N9 vs N7 alkylation) and stereoisomers, necessitating rigorous and costly purification steps such as repeated silica gel column chromatography. Literature reports indicate yields for these direct openings often hover between 27% and 50%, creating a significant material loss at a late stage of synthesis. Furthermore, the amino group on the guanine base requires protection and subsequent deprotection, adding unnecessary synthetic steps and introducing instability, as protected guanine intermediates are prone to decomposition on silica columns.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
The traditional reliance on guanine ring-opening creates a cascade of downstream processing challenges that severely impact commercial viability. The formation of diastereomeric mixtures during the alkylation step means that even after extensive purification, trace impurities can persist, complicating the regulatory approval process for the final Active Pharmaceutical Ingredient (API). The requirement for protecting groups on the guanine exocyclic amine adds complexity; for instance, MMT protection is difficult to manage and often requires chromatographic purification which leads to product decomposition. Additionally, alternative routes utilizing silane precursors for hydroxyl groups demand intensive oxidative conditions and strong acids or bases for conversion, which can degrade the sensitive nucleoside structure. These factors collectively result in a process that is not only low-yielding but also environmentally burdensome due to high solvent usage for chromatography.
The Novel Approach
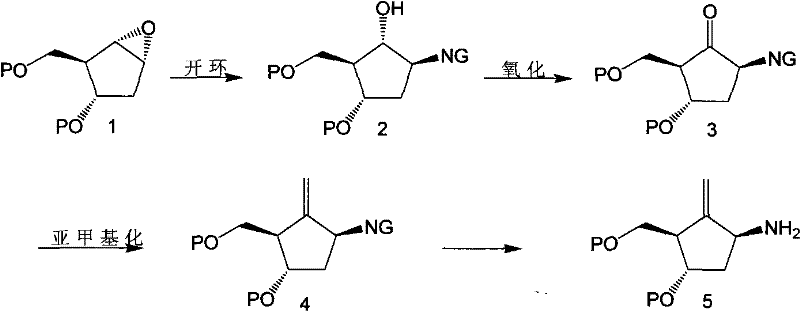
In stark contrast, the methodology described in CN101838207B adopts a "build-up" strategy where the carbocyclic ring is fully functionalized prior to nucleobase attachment. This route begins with the nucleophilic opening of a protected epoxy cyclopentane using nitrogenous reagents such as phthalimide or azides, rather than the bulky guanine base. This initial step sets the stereochemistry cleanly without the steric hindrance of the purine ring. Subsequent oxidation of the resulting hydroxyl group to a ketone, followed by methylenation to install the critical exocyclic double bond, proceeds with high efficiency. Only after the carbocyclic scaffold is complete is the nitrogen functionality converted to a primary amine and coupled with a pyrimidine derivative. This sequence effectively bypasses the regioselectivity issues of direct alkylation, as the cyclization to form the purine ring occurs intramolecularly under controlled acidic conditions, ensuring exclusive formation of the desired N9 linkage.
Mechanistic Insights into Carbocyclic Amine Construction and Purine Cyclization
The core of this synthetic breakthrough lies in the precise manipulation of the cyclopentane ring stereochemistry and functional groups. The process initiates with the ring-opening of a protected epoxide, such as a benzyl-protected oxabicyclohexane, using a nitrogen nucleophile like potassium phthalimide in a polar aprotic solvent like DMF. This SN2-type opening occurs with inversion of configuration, establishing the correct stereochemistry at the C1 position of the future nucleoside. Following this, the secondary hydroxyl group is oxidized to a ketone using mild yet effective oxidants like Dess-Martin periodinane, which avoids over-oxidation or epimerization. The subsequent methylenation step, utilizing reagents such as Nysted or Tebbe reagent in the presence of titanium tetrachloride at low temperatures (-78°C to room temperature), installs the exocyclic methylene group with high fidelity. This sequence ensures that the reactive alkene is in place before the sensitive purine ring is constructed, preventing side reactions that could occur if the alkene were present during earlier harsh conditions.
Impurity control is inherently built into this mechanistic pathway. By deferring the formation of the purine ring until the final stages, the synthesis avoids the generation of N7-isomers which are thermodynamically accessible during direct alkylation. The coupling of the carbocyclic amine with a substituted pyrimidine (e.g., 2-amino-5-nitro-4-chloropyrimidine) is highly regioselective due to the electronic activation of the pyrimidine ring. The subsequent cyclization using triethyl orthoformate and strong acid catalysis closes the imidazole ring efficiently. Any unreacted starting materials or side products from the coupling step are chemically distinct from the final fused bicyclic system, facilitating easier crystallization or extraction. This mechanistic design eliminates the need for the tedious resin chromatography mentioned in prior art, resulting in a final product profile that meets stringent pharmaceutical purity specifications with significantly reduced processing effort.
How to Synthesize Entecavir Intermediates Efficiently
The synthesis of Entecavir intermediates via this novel route involves a logical progression of functional group transformations that prioritize yield and stereochemical integrity. The process begins with the preparation of the key carbocyclic amine, followed by its condensation with a pyrimidine precursor and final cyclization. This approach allows for the isolation and characterization of stable intermediates, providing multiple quality control checkpoints before the final API is generated. The detailed standardized synthetic steps for implementing this high-efficiency route are outlined below.
- Perform nucleophilic ring opening of a protected epoxy cyclopentane using a nitrogenous reagent like phthalimide or azide to form a hydroxy-amine intermediate.
- Oxidize the secondary hydroxyl group to a ketone using reagents such as Dess-Martin periodinane, followed by methylenation to introduce the exocyclic double bond.
- Deprotect the nitrogen functionality to yield the primary amine, then couple with a substituted pyrimidine derivative and cyclize using orthoformate to form the purine ring.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the shift to this synthetic route offers tangible economic and operational benefits that extend beyond simple yield improvements. The elimination of low-yielding ring-opening steps and the removal of chromatographic purification requirements directly translate to reduced raw material consumption and lower waste disposal costs. By avoiding the use of unstable protected guanine intermediates, the process reduces the risk of batch failures and reprocessing, thereby enhancing overall supply reliability. The robustness of the reactions, which utilize standard reagents like Dess-Martin periodinane and orthoformates, ensures that the supply chain is not dependent on exotic or hard-to-source catalysts, mitigating the risk of raw material shortages.
- Cost Reduction in Manufacturing: The most significant cost driver in nucleoside synthesis is often the loss of material during purification and the expense of protecting group chemistry. This novel route drastically simplifies the process by removing the need for guanine protection and deprotection cycles. Furthermore, the avoidance of repeated silica gel column chromatography, which consumes vast quantities of solvents and adsorbents, leads to substantial cost savings in both materials and labor. The higher overall yield resulting from improved regioselectivity means that less starting material is required to produce the same amount of API, effectively lowering the cost of goods sold (COGS) for the final Entecavir product.
- Enhanced Supply Chain Reliability: Supply continuity is critical for antiviral medications, and this process enhances reliability by utilizing stable, isolable intermediates. The carbocyclic amine intermediate can be synthesized and stockpiled independently of the nucleobase coupling steps, allowing for flexible production scheduling. The reagents used, such as phthalimide and standard oxidants, are commodity chemicals with reliable global supply chains, reducing the vulnerability to single-source supplier issues. Additionally, the simplified workup procedures reduce the turnaround time between batches, enabling manufacturers to respond more quickly to fluctuations in market demand without compromising on quality.
- Scalability and Environmental Compliance: Scaling up chemical processes often reveals hidden inefficiencies, particularly those related to heat transfer and mixing in viscous chromatography columns. This synthetic route is inherently more scalable because it relies on solution-phase reactions and crystallization-based purifications rather than column chromatography. The reduction in solvent usage aligns with green chemistry principles, lowering the environmental footprint of the manufacturing process. This not only simplifies regulatory compliance regarding waste discharge but also reduces the energy costs associated with solvent recovery and distillation, making the process economically and environmentally sustainable for large-scale commercial production.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Entecavir synthesis method. These answers are derived directly from the experimental data and comparative analysis provided in the patent literature, offering clarity on yield expectations, purity profiles, and operational feasibility for potential manufacturing partners.
Q: Why is this new synthetic route superior to traditional guanine ring-opening methods?
A: Traditional methods involve direct ring-opening of guanine derivatives with epoxy cyclopentanes, which often suffer from low yields (27-50%) and difficult separation of 9-N vs 7-N isomers. This novel route constructs the purine ring after the carbocyclic sugar is fully formed, ensuring high regioselectivity and eliminating the need for complex chromatographic purification of isomers.
Q: What are the key advantages regarding impurity control in this process?
A: The process avoids the formation of stereoisomeric mixtures common in direct alkylation. By using a protected amine intermediate (such as phthalimide) that is only deprotected after the exocyclic double bond is installed, the synthesis prevents side reactions on the nucleobase. Furthermore, the final cyclization step is highly specific, leading to a final product with significantly higher purity without requiring resin chromatography.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the method is designed for industrial suitability. It replaces harsh conditions and unstable intermediates found in older patents with robust reactions like Dess-Martin oxidation and Nysted methylenation. The elimination of repeated silica gel column chromatography steps drastically reduces solvent consumption and processing time, making it highly scalable for commercial API manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Entecavir Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and robust infrastructure. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated chemistry described in CN101838207B can be seamlessly translated into industrial reality. We maintain stringent purity specifications and operate rigorous QC labs equipped to handle the complex analytical requirements of nucleoside intermediates, guaranteeing that every batch meets the exacting standards required for antiviral API manufacturing.
We invite global pharmaceutical partners to collaborate with us to leverage this advanced synthetic technology for your Entecavir supply needs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this route can optimize your budget. Please contact us to request specific COA data for our intermediates and comprehensive route feasibility assessments, and let us help you secure a competitive advantage in the antiviral market through superior chemical manufacturing.