Advanced Synthesis of Phenyl-Substituted Artemisinin Derivatives for Commercial API Production
Advanced Synthesis of Phenyl-Substituted Artemisinin Derivatives for Commercial API Production
The pharmaceutical landscape is constantly evolving, driven by the need for more effective antimalarial and antiviral agents with improved pharmacokinetic profiles. Patent CN1049435C introduces a groundbreaking class of artemisinin derivatives containing phenyl and heterocyclic radicals, offering a robust pathway for the development of next-generation therapeutic intermediates. This technology leverages the potent sesquiterpene lactone scaffold of artemisinin, modifying it through precise acid-catalyzed reactions to enhance biological activity against resistant strains of malaria and even exhibiting potential against HIV and tumors. For R&D directors and procurement specialists seeking reliable pharmaceutical intermediates suppliers, understanding the nuances of this synthesis is critical for securing a competitive edge in the global API market.
The core innovation lies in the versatility of the substitution pattern, allowing for the attachment of diverse aromatic and heterocyclic groups to the dihydroartemisinin backbone. This structural flexibility is paramount for optimizing drug efficacy and reducing toxicity, addressing key pain points in modern drug discovery. By utilizing dihydroartemisinin compounds as the starting material, the process ensures a direct lineage to the natural product's proven efficacy while overcoming limitations associated with the parent compound's stability and solubility. The following analysis delves deep into the mechanistic advantages and commercial viability of this patented methodology.
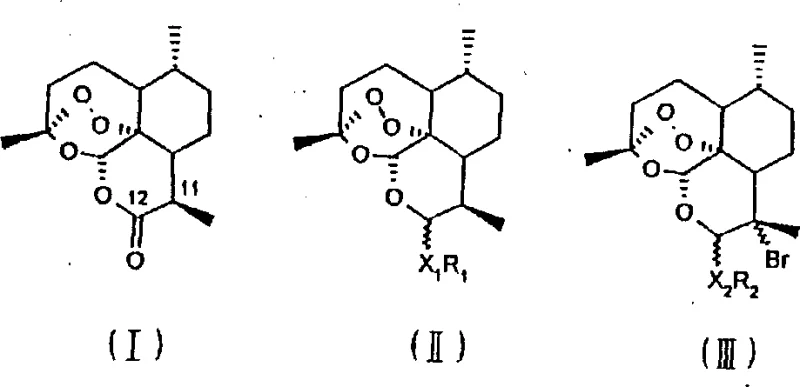
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional methods for modifying the artemisinin scaffold often suffer from harsh reaction conditions that can degrade the sensitive endoperoxide bridge, which is essential for antimalarial activity. Many prior art processes require extreme temperatures or strong bases that lead to significant decomposition of the starting material, resulting in low overall yields and complex purification challenges. Furthermore, conventional routes frequently lack the regioselectivity needed to introduce bulky phenyl or heterocyclic groups at the desired positions without affecting other sensitive functional groups on the molecule. This lack of precision often necessitates multiple protection and deprotection steps, drastically increasing the cost of goods sold and extending the lead time for high-purity pharmaceutical intermediates.
Additionally, older synthetic pathways often rely on expensive or difficult-to-source reagents that complicate the supply chain continuity. The inability to easily scale these processes due to safety concerns regarding exothermic reactions or the handling of unstable intermediates poses a significant risk for commercial scale-up of complex polymer additives or API precursors. Consequently, manufacturers face bottlenecks in production capacity and struggle to meet the stringent purity specifications required by regulatory bodies for clinical-grade materials. These inefficiencies highlight the urgent need for a more streamlined and robust synthetic approach.
The Novel Approach
The methodology described in CN1049435C represents a paradigm shift by employing mild acidic catalysis to drive the substitution reaction efficiently. By reacting dihydroartemisinin or its esters with phenyl amines, phenols, or heterocyclic compounds in the presence of catalysts like boron trifluoride ethyl ether complex or tin tetrachloride, the process achieves high conversion rates under gentle thermal conditions ranging from -10°C to 40°C. This approach preserves the integrity of the critical endoperoxide bridge while successfully installing the desired aromatic moieties. The use of common polar solvents such as methylene dichloride, acetonitrile, or tetrahydrofuran further simplifies the operational protocol, making it highly amenable to large-scale manufacturing environments.
Moreover, this novel route eliminates the need for cumbersome protection strategies, as the acidic conditions selectively activate the hemiacetal hydroxyl group of dihydroartemisinin for nucleophilic attack. The workup procedure is equally straightforward, involving simple aqueous washes and silica gel column chromatography to isolate the pure product. This simplicity translates directly into reduced operational complexity and lower waste generation, aligning with green chemistry principles. For procurement managers, this means a more predictable supply chain with fewer variables that could disrupt production schedules, ensuring a steady flow of high-quality intermediates for downstream drug formulation.
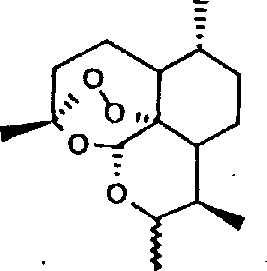
Mechanistic Insights into Acid-Catalyzed Substitution
The reaction mechanism centers on the activation of the anomeric carbon at the C-10 position of the dihydroartemisinin ring system by the Lewis or Brønsted acid catalyst. Upon coordination with the catalyst, the hydroxyl group becomes a better leaving group, generating a transient oxocarbenium ion intermediate that is highly electrophilic. This activated species is then susceptible to nucleophilic attack by the electron-rich aromatic rings of the added amines or phenols. The stereochemical outcome of this reaction can be influenced by the choice of solvent and catalyst, allowing for the selective formation of either alpha or beta isomers, as indicated by the wave and straight line notations in the structural formulas. This level of control is essential for producing single-isomer drugs with consistent therapeutic profiles.
Impurity control is inherently built into this mechanism due to the specificity of the acid-catalyzed pathway. Unlike radical-based modifications that can lead to random fragmentation of the sesquiterpene skeleton, this ionic mechanism targets a specific site, minimizing the formation of side products. The patent data demonstrates that by carefully monitoring the reaction progress via thin-layer chromatography, operators can quench the reaction precisely upon completion, preventing over-reaction or degradation. The subsequent purification via silica gel chromatography effectively removes any unreacted starting materials or minor byproducts, ensuring the final product meets the rigorous purity standards demanded by the pharmaceutical industry. This mechanistic clarity provides R&D teams with the confidence to optimize the process for maximum efficiency.
How to Synthesize Artemisinin Derivatives Efficiently
The synthesis of these valuable intermediates follows a logical and reproducible sequence that balances reaction kinetics with product stability. The process begins with the dissolution of the dihydroartemisinin precursor in a suitable anhydrous polar solvent, creating a homogeneous reaction medium. The nucleophile, whether it be an aniline derivative or a heterocyclic base, is then introduced along with a catalytic amount of the chosen acid. Maintaining the temperature within the specified range is crucial to manage the exotherm and ensure selectivity. Detailed standardized synthesis steps see the guide below.
- Dissolve dihydroartemisinin or its esters in a polar solvent such as methylene dichloride or acetonitrile.
- Add the chosen nucleophile (phenyl amine, phenol, or heterocyclic compound) and an acidic catalyst like boron trifluoride ethyl ether complex.
- Stir the reaction mixture at controlled temperatures between -10°C and 40°C, then purify the product via silica gel column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For supply chain heads and procurement managers, the adoption of this synthetic route offers substantial strategic benefits beyond mere technical feasibility. The reliance on commodity chemicals like dihydroartemisinin and common organic solvents significantly mitigates the risk of raw material shortages. Since the starting materials are derived from abundant natural sources or established synthetic supply chains, the volatility associated with exotic reagents is eliminated. This stability allows for long-term contracting and better budget forecasting, which is essential for maintaining healthy margins in the competitive generic drug market. The robustness of the process also means that technology transfer between manufacturing sites can be executed with minimal friction.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the avoidance of cryogenic conditions drastically simplify the infrastructure requirements for production. By operating at near-ambient temperatures, energy consumption for heating and cooling is minimized, leading to significant operational expenditure savings. Furthermore, the high atom economy of the substitution reaction means that less raw material is wasted, directly lowering the cost per kilogram of the final active pharmaceutical ingredient. The simplified workup procedure reduces the volume of solvents required for purification, contributing to lower waste disposal costs and a smaller environmental footprint.
- Enhanced Supply Chain Reliability: The use of widely available catalysts such as phosphoric acid or tosic acid ensures that production is never halted due to the unavailability of specialized reagents. This redundancy in catalyst selection provides a buffer against supply chain disruptions, ensuring continuous operation even if one supplier faces issues. The scalability of the reaction from gram to multi-kilogram scales has been demonstrated in the patent examples, proving that the chemistry holds up under increased load. This reliability is critical for meeting tight delivery windows and maintaining trust with downstream pharmaceutical partners who depend on just-in-time inventory models.
- Scalability and Environmental Compliance: The process generates minimal hazardous byproducts, primarily consisting of aqueous salt solutions that are easy to treat and dispose of in accordance with environmental regulations. The absence of heavy metals in the catalyst system simplifies the purification process and ensures that the final product is free from toxic metal residues, a common regulatory hurdle in API manufacturing. This compliance advantage accelerates the regulatory approval process for new drug applications. Additionally, the ability to recycle solvents like methylene dichloride or ethyl acetate further enhances the sustainability profile of the manufacturing process, aligning with corporate social responsibility goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these artemisinin derivatives. The answers are derived directly from the experimental data and claims presented in the patent documentation, providing a factual basis for decision-making. Understanding these details helps stakeholders assess the fit of this technology within their existing portfolios and identify potential areas for collaboration or licensing.
Q: What catalysts are suitable for synthesizing these artemisinin derivatives?
A: The patent specifies a wide range of acidic catalysts including boron trifluoride ethyl ether complex, tin tetrachloride, titanium tetrachloride, trifluoroacetic acid, and trifluoromethanesulfonic acid trimethylsilyl group (TMSOTf).
Q: What are the biological activities of these new derivatives?
A: Preliminary pharmacological screening indicates these derivatives possess significant protozoacide effects, immunoregulation capabilities, and antiviral activities including anti-AIDS virus properties, which the parent artemisinin compound lacks.
Q: Can this process be scaled for industrial production?
A: Yes, the process utilizes readily available raw materials like dihydroartemisinin and common organic solvents, with mild reaction conditions that facilitate safe and scalable commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Artemisinin Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the immense potential of these phenyl-substituted artemisinin derivatives in combating resistant diseases and expanding the therapeutic utility of this classic natural product. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from the laboratory bench to full-scale manufacturing. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of intermediate delivered meets the highest international standards for safety and efficacy.
We invite you to engage with our technical procurement team to discuss how this patented technology can be integrated into your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this more efficient synthetic route. We are ready to provide specific COA data and route feasibility assessments tailored to your unique production requirements, helping you secure a reliable source of high-quality pharmaceutical intermediates for your next generation of life-saving medicines.