Advanced Bilastine Synthesis: Overcoming Hydrolysis Defects for Commercial Scale-up
Advanced Bilastine Synthesis: Overcoming Hydrolysis Defects for Commercial Scale-up
The pharmaceutical industry constantly seeks robust synthetic routes that minimize impurity profiles while maximizing yield, particularly for high-volume antihistamines like Bilastine. A significant breakthrough in this domain is detailed in patent CN112110893A, which outlines a novel preparation method that fundamentally alters the sequence of functional group installation. Unlike traditional approaches that risk structural degradation during late-stage deprotection, this invention utilizes a stable carboxylic acid intermediate to secure the molecular scaffold before introducing the sensitive ether linkage. This strategic pivot not only enhances the chemical purity of the final Active Pharmaceutical Ingredient (API) but also streamlines the manufacturing workflow, offering a compelling value proposition for reliable API supplier partnerships aiming for cost reduction in pharmaceutical manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
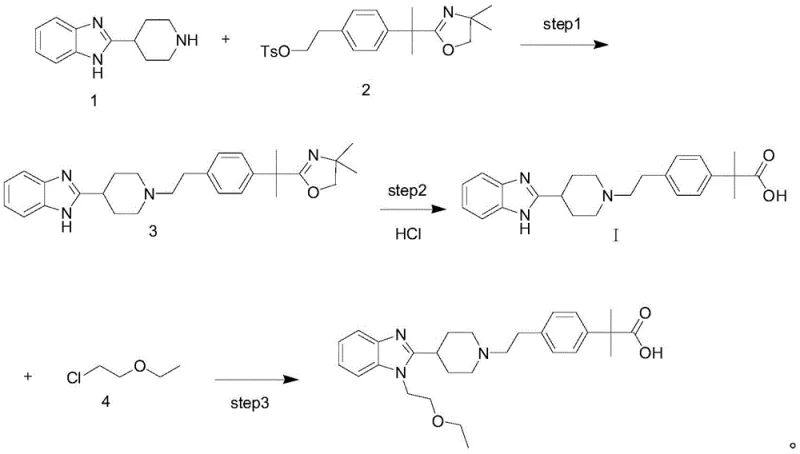
The Limitations of Conventional Methods
Historically, the synthesis of Bilastine has been plagued by a specific chemical vulnerability associated with the timing of ether bond formation. In prior art methodologies, such as those described in patent CN1105716C, the synthetic route often involves the construction of the core piperidine-benzimidazole structure followed by the hydrolysis of an oxazole protecting group. The critical flaw in this sequence is that the ether moiety is typically installed prior to this hydrolysis step. Consequently, when strong acids or bases are employed to cleave the oxazole ring, these harsh conditions frequently attack the adjacent ether bond. This side reaction leads to the cleavage of the ethoxy-ethyl group, generating alcohol impurities that possess physicochemical properties nearly identical to the target molecule. As illustrated in the background analysis of the patent, these alcohol byproducts are notoriously difficult to separate via standard crystallization or chromatography, severely impacting the overall quality and safety profile of the medicine.
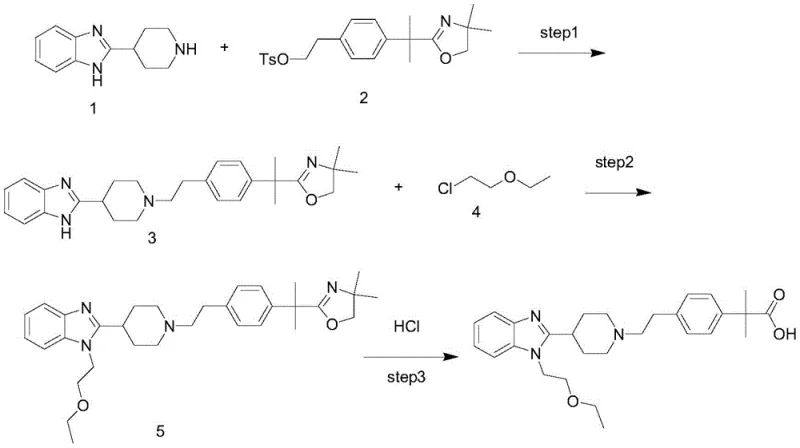
The Novel Approach
The innovative strategy presented in the disclosed patent circumvents this degradation pathway by reversing the order of operations. Instead of hydrolyzing a protected group after etherification, the new method prioritizes the formation of the key carboxylic acid intermediate, designated as Formula I. By establishing the acid functionality first—either through the hydrolysis of an oxazole precursor or a methyl ester—the synthesis ensures that the sensitive ether bond is not present during the harsh deprotection phase. Once the pure acid intermediate is isolated, the final step involves a mild nucleophilic substitution to attach the 2-ethoxyethyl group. This approach effectively decouples the harsh hydrolysis conditions from the sensitive ether linkage, virtually eliminating the formation of the problematic alcohol impurities and resulting in a much cleaner crude product that requires less intensive purification.
Mechanistic Insights into Nucleophilic Substitution and Hydrolysis
The core of this improved synthesis lies in the precise control of nucleophilicity and electrophilicity across three distinct stages. Initially, the coupling reaction between 2-piperidin-4-yl-1H-benzimidazole and the phenyl-ethyl derivative relies on the nucleophilic attack of the piperidine nitrogen. Whether using a tosylate leaving group on the phenyl side or an activated ester, the reaction is driven by base catalysis, typically using sodium carbonate in polar aprotic solvents like DMF. Following this, the hydrolysis step is critical; if starting from the oxazole variant, acidic conditions (e.g., HCl reflux) are used to open the ring without affecting the already stable acid or ester groups elsewhere in the molecule. If starting from the methyl ester, alkaline hydrolysis with sodium hydroxide efficiently converts the ester to the free acid. The final mechanistic highlight is the etherification of the benzimidazole nitrogen. Here, sodium hydride acts as a strong, non-nucleophilic base to deprotonate the benzimidazole NH, generating a reactive anion that attacks 2-chloroethyl ether in an SN2 fashion.
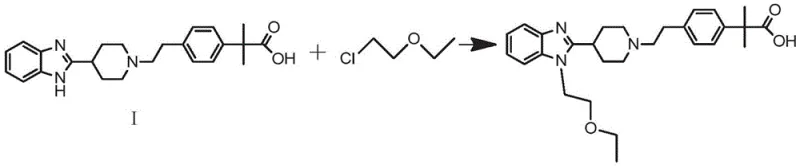
From an impurity control perspective, this mechanism offers superior selectivity. In the conventional route, the competition between oxazole hydrolysis and ether cleavage is a kinetic nightmare, often requiring precise temperature control that is hard to maintain on a multi-ton scale. In the new route, the hydrolysis step targets only the oxazole or ester functionality, leaving the rest of the molecule inert. Furthermore, the final alkylation step is highly specific to the benzimidazole nitrogen due to the pKa difference between the benzimidazole NH and the carboxylic acid (which is managed by stoichiometry or protection/deprotection logic inherent in the salt forms). This orthogonality ensures that side reactions are minimized, leading to a simpler impurity profile that is easier to characterize and control, a crucial factor for regulatory approval and batch consistency.
How to Synthesize Bilastine Efficiently
The synthesis of Bilastine via this optimized route is designed for operational simplicity and high throughput. The process generally follows a linear three-step sequence that can be executed in standard stainless steel reactors without the need for specialized high-pressure or cryogenic equipment. The initial coupling establishes the carbon-nitrogen backbone, followed by a hydrolysis step that reveals the critical carboxylic acid handle. The final step installs the pharmacophore-defining ether chain. For process chemists looking to implement this, the detailed standardized synthesis steps are outlined below, providing a clear roadmap from raw materials to the final API.
- Coupling Reaction: React 2-piperidin-4-yl-1H-benzimidazole with a protected phenyl-ethyl derivative (either oxazole or ester form) in DMF with sodium carbonate at elevated temperatures.
- Hydrolysis: Convert the protected intermediate into the key carboxylic acid (Formula I) using acidic or alkaline hydrolysis conditions, ensuring the ether bond remains intact.
- Final Etherification: Alkylate the benzimidazole nitrogen of the acid intermediate with 2-chloroethyl ether using sodium hydride in DMF to yield Bilastine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the shift to this novel synthetic route translates directly into risk mitigation and cost efficiency. The primary economic driver here is the drastic reduction in post-reaction processing. By eliminating the formation of structurally similar alcohol impurities, the need for repetitive recrystallizations or expensive preparative HPLC purification is significantly reduced. This not only lowers the consumption of solvents and adsorbents but also shortens the cycle time per batch, allowing for higher throughput in existing manufacturing facilities. Furthermore, the raw materials required, such as 2-chloroethyl ether and simple benzimidazole derivatives, are commodity chemicals with stable global supply chains, reducing the risk of raw material shortages that often plague complex custom syntheses.
- Cost Reduction in Manufacturing: The elimination of difficult-to-remove impurities directly impacts the bottom line by improving the overall yield of the process. In traditional methods, yield losses occur not just from side reactions but from the material lost during aggressive purification attempts to meet strict impurity limits. By preventing the impurity from forming in the first place, the new method preserves mass balance. Additionally, the avoidance of transition metal catalysts or exotic reagents means that waste disposal costs are lower, and the process is more environmentally sustainable, aligning with modern green chemistry mandates without compromising economic viability.
- Enhanced Supply Chain Reliability: The robustness of this synthesis enhances supply security. Because the reaction conditions are mild (typically ranging from 45°C to 80°C) and do not require stringent anhydrous environments for every step, the process is less susceptible to minor variations in utility quality or operator error. This reliability ensures consistent batch-to-batch quality, which is essential for maintaining long-term supply contracts with major pharmaceutical companies. The use of common solvents like ethanol and DMF further ensures that solvent supply disruptions are unlikely to halt production.
- Scalability and Environmental Compliance: Scaling up chemical processes often introduces thermal hazards, but this route operates well within safe thermal limits. The exotherms associated with the base-mediated steps are manageable, and the hydrolysis steps utilize aqueous workups that are easily handled in standard separation units. From an environmental standpoint, the reduced solvent usage and the absence of heavy metal contaminants simplify the wastewater treatment process. This makes the technology easier to permit in regions with strict environmental regulations, facilitating the establishment of diversified manufacturing sites to ensure business continuity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Bilastine synthesis technology. These answers are derived directly from the experimental data and technical specifications provided in the patent literature, ensuring that stakeholders have accurate information for decision-making. Understanding these nuances is critical for evaluating the feasibility of technology transfer and the potential return on investment for adopting this newer methodology.
Q: Why does the conventional Bilastine synthesis generate difficult-to-remove impurities?
A: Conventional methods often introduce the ether group early and then perform a harsh hydrolysis on an oxazole ring. This strong acid or base treatment frequently cleaves the sensitive ether bond, generating alcohol impurities with structures very similar to the final product, making purification extremely difficult.
Q: What is the key advantage of using Formula I as an intermediate?
A: Using the carboxylic acid intermediate (Formula I) allows the etherification step to occur as the final transformation. This avoids exposing the ether bond to harsh hydrolysis conditions, thereby preventing the formation of alcohol byproducts and significantly simplifying downstream purification.
Q: Is this synthesis route suitable for large-scale manufacturing?
A: Yes, the route utilizes economically available raw materials, operates at mild temperatures (45-80°C), and avoids complex transition metal catalysts. The high yields reported (averaging over 80% per step) and simple workup procedures make it highly scalable for industrial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Bilastine Supplier
The technical advantages of this synthesis route highlight the importance of partnering with a manufacturer that possesses deep process chemistry expertise. NINGBO INNO PHARMCHEM stands at the forefront of this capability, leveraging extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facility is equipped to handle the specific unit operations required for this synthesis, including efficient DMF recovery systems and precise pH control for the hydrolysis steps. We maintain stringent purity specifications and operate rigorous QC labs to ensure that every batch of Bilastine intermediate or API meets the highest international standards, providing our partners with the confidence needed for successful drug registration and market launch.
We invite procurement leaders and R&D heads to engage with us for a Customized Cost-Saving Analysis tailored to your specific volume requirements. By optimizing the supply chain for this critical antihistamine, we can help you achieve significant operational efficiencies. Please contact our technical procurement team to request specific COA data and route feasibility assessments, and let us demonstrate how our advanced manufacturing capabilities can support your long-term strategic goals.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →