Advanced Purification Technology for Pravastatin Sodium: Ensuring Clinical Safety and Commercial Scalability
The pharmaceutical landscape for lipid-lowering agents demands uncompromising standards regarding purity and safety, particularly for widely prescribed statins like Pravastatin Sodium. Patent CN102070447B introduces a transformative preparation method that addresses critical limitations in existing purification technologies, offering a pathway to superior product quality. This innovation leverages a sophisticated combination of strong-acid ion exchange resin adsorption and controlled mild elution to isolate Pravastatin Sodium with exceptional precision. Unlike conventional crystallization techniques that often struggle with residual impurities, this approach ensures the removal of toxic byproducts and heavy metals while preserving the delicate stereochemistry of the molecule. For R&D directors and procurement specialists, understanding this technological leap is essential for securing a reliable supply of high-purity API intermediates. The method not only enhances clinical safety by reducing side effects associated with impurities but also streamlines the manufacturing workflow, presenting a compelling case for adoption in large-scale commercial production environments where consistency is paramount.
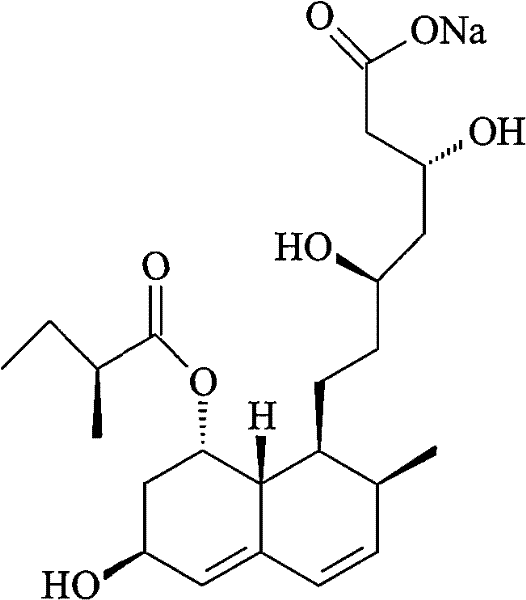
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the purification of SQ-31000, commonly known as Pravastatin Sodium, has relied heavily on complex acid-base reactions followed by organic solvent extraction and crystallization. These traditional methodologies, such as those referenced in prior art like CN08820812, are fraught with operational inefficiencies and chemical risks. The primary drawback lies in the harsh conditions required to adjust pH levels, which often necessitate the use of strong alkaline reagents. These aggressive chemical environments pose a significant threat to the structural integrity of the pravastatin molecule, specifically targeting the vulnerable ester linkage. When subjected to strong bases, the ester group is prone to hydrolysis, leading to the formation of unwanted degradation products that drastically lower the overall yield and complicate downstream purification. Furthermore, the reliance on extensive organic solvent usage increases both the environmental footprint and the operational cost, creating bottlenecks in waste management and solvent recovery that hinder scalable manufacturing efforts.
The Novel Approach
In stark contrast, the novel preparation method detailed in the patent data circumvents these pitfalls through a gentle yet highly effective ion exchange mechanism. By utilizing a strongly acidic ion exchange resin, the process converts the sodium salt into its corresponding acid form without exposing the molecule to destructive alkaline conditions. This strategic conversion allows for the selective adsorption of the target compound while leaving many impurities in the solution or binding them differently. The subsequent elution step employs a mild aqueous solution of sodium carbonate or sodium bicarbonate at low concentrations, typically between 0.1% and 2%. This mild elution regime is crucial as it regenerates the sodium salt form of pravastatin under near-neutral conditions, effectively eliminating the risk of ester hydrolysis. The result is a purification trajectory that maintains high yields while achieving purity levels that surpass traditional methods, offering a robust solution for the cost reduction in pharmaceutical intermediate manufacturing.
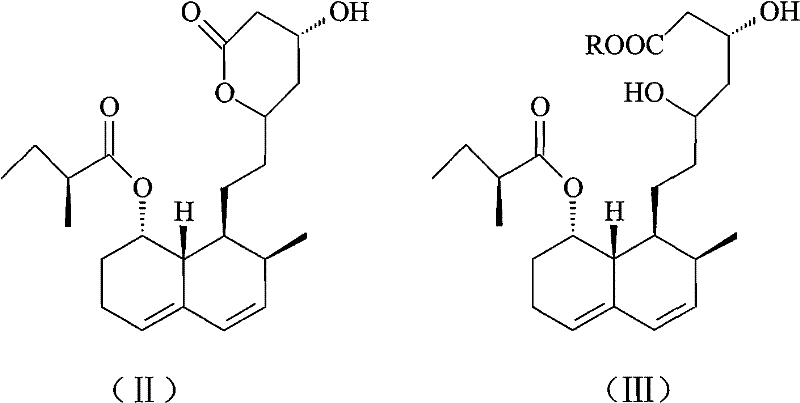
Mechanistic Insights into Ion Exchange and Chromatographic Separation
The core of this technological advancement lies in the precise manipulation of ionic interactions within the resin matrix. When the crude Pravastatin Sodium solution passes through the strong-acid cation exchange resin, the sodium ions associated with the carboxylate group are exchanged for hydrogen ions provided by the sulfonic acid groups on the resin. This transforms the soluble sodium salt into pravastatin acid, which exhibits different adsorption characteristics and binds tightly to the resin matrix. This step is not merely a filtration process but a chemical transformation that stabilizes the molecule against hydrolysis. The resin acts as a selective barrier, capturing the target acid while allowing various inorganic salts and polar impurities to pass through or be washed away. The specificity of this interaction is governed by the cross-linked polymeric structure of the resin, which provides a high surface area for interaction while maintaining mechanical stability under flow conditions, ensuring that the process remains viable for continuous industrial operations.
Following the adsorption phase, the control of the elution environment becomes the critical determinant of final purity. The use of sodium bicarbonate or sodium carbonate serves a dual purpose: it provides the necessary sodium ions to reform the pravastatin sodium salt and generates carbon dioxide as a byproduct, which naturally vents from the system without introducing new ionic contaminants. This is followed by a rigorous silica gel column chromatography step, utilizing a mobile phase of acetonitrile and water. This final polishing stage exploits the subtle differences in polarity between the pravastatin sodium and any remaining trace impurities. The stationary phase, typically silica gel with a specific particle diameter range of 60-300 μm, ensures high resolution separation. By optimizing the flow rate and pressure, the process achieves a sharp separation profile, collecting only the fractions that meet the stringent purity specifications required for high-purity OLED material or pharmaceutical applications, thereby guaranteeing a product free from genotoxic impurities.
How to Synthesize Pravastatin Sodium Efficiently
The implementation of this synthesis route requires careful attention to the operational parameters defined in the patent embodiments to ensure reproducibility and optimal yield. The process begins with the dissolution of the crude bulk drug in water, followed by passage through a fixed bed of D001 type strongly acidic styrene cation exchange resin. The contact time is critical, generally maintained around 1.5 hours to ensure complete ion exchange equilibrium. Once the acid form is adsorbed, the column is eluted with a carefully prepared 0.5% to 1% sodium bicarbonate solution at a controlled flow rate of approximately 1.0 ml/min. This slow elution ensures complete conversion back to the sodium salt without localized pH spikes that could damage the product. The resulting eluate is then subjected to silica gel chromatography using an acetonitrile-water mixture, followed by drying and concentration. Detailed standardized synthesis steps see the guide below.
- Adsorb crude Pravastatin Sodium onto a strongly acidic ion exchange resin to convert it into pravastatin acid form, effectively trapping impurities.
- Elute the resin using a mild 0.1% to 2% sodium bicarbonate or sodium carbonate aqueous solution to regenerate the sodium salt without hydrolyzing the ester group.
- Perform final separation and purification via silica gel column chromatography using an acetonitrile-water mobile phase to isolate the pure product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this purification technology translates directly into enhanced operational resilience and cost efficiency. The elimination of harsh chemical reagents and the reduction in solvent consumption significantly lower the variable costs associated with raw material procurement and waste disposal. By avoiding the degradation of the product during purification, the overall yield is substantially improved, meaning less starting material is required to produce the same amount of finished API. This efficiency gain is critical in a market where raw material prices can be volatile. Furthermore, the simplicity of the process reduces the complexity of the manufacturing equipment needed, lowering capital expenditure requirements for scale-up. The ability to consistently produce material with purity exceeding 99.9% minimizes the risk of batch rejection, ensuring a smoother flow of goods through the supply chain and reducing the lead time for high-purity pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The process fundamentally alters the cost structure by removing the need for expensive solvent recovery systems associated with heavy organic extraction. Since the primary medium is water and mild buffers, the energy consumption for solvent evaporation is drastically reduced. Additionally, the prevention of ester hydrolysis means that the yield loss typically seen in alkaline purification is avoided, directly improving the cost-per-kilogram of the final product. This qualitative improvement in process efficiency allows manufacturers to offer more competitive pricing without compromising on margin, providing a distinct advantage in tender negotiations for generic drug production.
- Enhanced Supply Chain Reliability: Reliability in the supply of critical statin intermediates is paramount for global pharmaceutical companies. This method utilizes commercially available resins and common chemicals, reducing the risk of supply disruptions caused by specialty reagent shortages. The robustness of the ion exchange process allows for flexible production scheduling, as the resin columns can be regenerated and reused multiple times without significant loss of performance. This durability ensures a continuous production capability, mitigating the risks of stockouts and enabling suppliers to meet sudden surges in demand with greater agility and confidence.
- Scalability and Environmental Compliance: As regulatory pressures regarding environmental discharge intensify, this technology offers a greener alternative to traditional synthesis. The reduction in organic solvent usage and the absence of heavy metal catalysts simplify the wastewater treatment process, ensuring compliance with strict environmental regulations. The process is inherently scalable from laboratory benchtop to multi-ton commercial production, as the principles of ion exchange and chromatography scale linearly with column dimensions. This scalability ensures that the quality established during development is maintained during commercial scale-up of complex pharmaceutical additives, facilitating a seamless transition from clinical trials to full-market availability.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this purification technology. They are derived from the specific advantages outlined in the patent documentation and are intended to clarify the operational benefits for potential partners. Understanding these details is crucial for evaluating the feasibility of integrating this method into existing production lines. The answers reflect the consensus on best practices for handling ion exchange resins and chromatographic systems in a GMP environment.
Q: How does this purification method prevent ester hydrolysis compared to traditional alkaline treatments?
A: Traditional methods often use strong bases like sodium hydroxide which can attack the sensitive ester group in the pravastatin molecule. This patented method utilizes a strong-acid ion exchange resin to first convert the salt to its acid form, followed by elution with mild weak bases like sodium bicarbonate. This controlled pH environment prevents the hydrolysis of the ester bond, thereby maintaining molecular integrity and significantly improving overall yield and purity.
Q: What level of purity can be achieved using this ion exchange and chromatography protocol?
A: The described process is capable of achieving exceptional purity levels exceeding 99.9%. By combining the selective adsorption capabilities of the macroporous strong-acid resin with precise silica gel column chromatography, the method effectively removes bacterial endotoxins, heavy metals, and organic impurities that are typically difficult to eliminate in standard crystallization processes.
Q: Is this purification technology suitable for large-scale industrial production?
A: Yes, the process is explicitly designed for industrial scalability. It utilizes commercially available resins and common solvents like water and acetonitrile. The operation can be adapted to fixed-bed or continuous flow systems, allowing for significant throughput increases while maintaining consistent quality control standards required for commercial API manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Pravastatin Sodium Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent theory to commercial reality requires a partner with deep technical expertise and robust manufacturing capabilities. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the promising results of CN102070447B can be realized on an industrial scale. We maintain stringent purity specifications across all our product lines, supported by rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our commitment to quality ensures that every batch of Pravastatin Sodium meets the exacting standards required by global regulatory bodies, providing you with a secure foundation for your drug development and manufacturing programs.
We invite you to collaborate with us to optimize your supply chain and reduce your overall production costs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality needs. By leveraging our advanced purification technologies, we can help you achieve significant efficiencies in your manufacturing process. We encourage you to contact us today to request specific COA data and route feasibility assessments, allowing you to make informed decisions that drive value for your organization and ensure the uninterrupted supply of critical medications to patients worldwide.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →