Advanced Manufacturing of BIBR-953 Antithrombin Inhibitors via Optimized Benzimidazole Synthesis
The global demand for effective anticoagulant therapies continues to drive innovation in the synthesis of direct thrombin inhibitors, particularly non-peptide variants that offer superior oral bioavailability. Patent CN1861596A discloses a groundbreaking methodology for synthesizing BIBR-953 and its prodrug BIBR-1048, which are second-generation antithrombin inhibitors developed to overcome the limitations of heparin and warfarin. This patent introduces a novel synthetic pathway that utilizes 3-nitro-4-chlorobenzoic acid as a cost-effective starting material, diverging significantly from prior art routes that relied on more expensive precursors. The technical breakthrough lies in the optimization of key reaction steps, including the replacement of problematic solvents and the implementation of high-efficiency coupling agents, resulting in a total yield approaching 50%. For pharmaceutical manufacturers, this represents a critical advancement in the commercial scale-up of complex pharmaceutical intermediates, ensuring a more robust supply chain for these vital cardiovascular therapeutics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this innovation, the synthesis of BIBR-953, as reported by Boehringer Ingelheim Pharma KG, relied on a route designated as Route (I) which presented significant industrial bottlenecks. The conventional method initiated with a relatively expensive raw material, Compound 1, and utilized N,N-dimethylformamide (DMF) as a solvent for the synthesis of the acyl chloride intermediate, Compound 2. The use of DMF is notoriously difficult in large-scale operations due to its high boiling point and challenging removal during workup, often requiring extensive aqueous washing and energy-intensive distillation. Furthermore, the cyclization step to form the benzimidazole core utilized CDI (carbonyldiimidazole), which, while effective on a small scale, proved less efficient in terms of yield and cost for bulk manufacturing. The subsequent amidination step involved acidolysis followed by treatment with ammonium carbonate, a two-step process that resulted in low yields and complicated purification protocols. Collectively, these inefficiencies culminated in a dismal overall yield of merely 11.1%, rendering the process economically unviable for cost reduction in API manufacturing on a commercial scale.
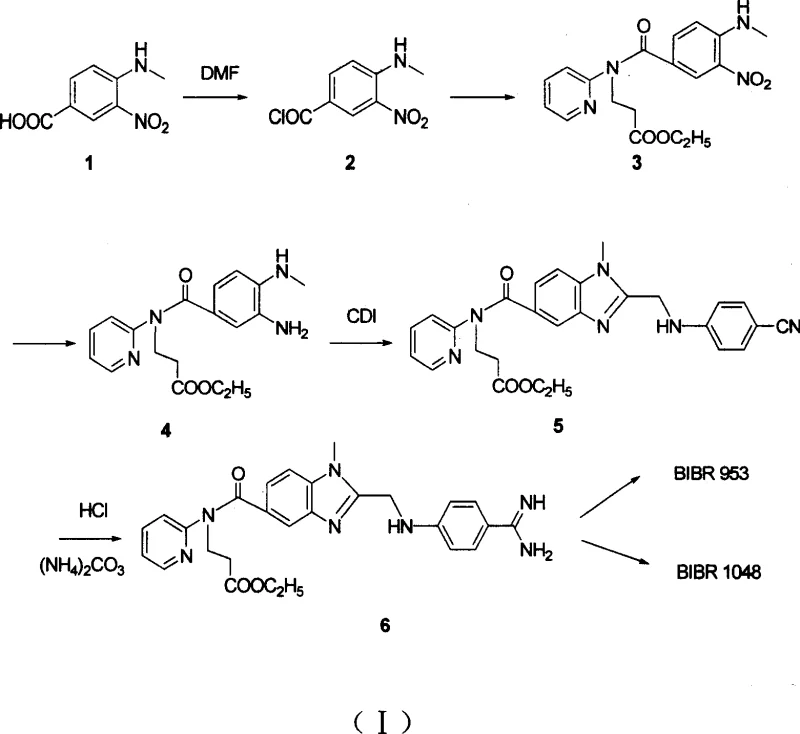
The Novel Approach
In stark contrast, the methodology described in CN1861596A revolutionizes the production landscape by introducing a streamlined, high-yield pathway starting from the廉价 and readily available 3-nitro-4-chlorobenzoic acid (Compound 13). A pivotal improvement is the substitution of DMF with dichloromethane (CH2Cl2) for the conversion of the carboxylic acid to the acyl chloride, which drastically simplifies the post-reaction processing and solvent recovery. The synthesis employs ethyl 3-(pyridin-2-ylimino)propanoate (Compound 10) and 1-(4-cyanophenylimino)acetic acid (Compound 12), both of which are synthesized from inexpensive precursors via efficient Michael addition and alkylation reactions respectively. The core benzimidazole ring closure is achieved using a superior coupling system comprising HOBT and EDCI, which enhances reaction kinetics and purity profiles compared to the traditional CDI method. Additionally, the amidination of the nitrile group is optimized by direct treatment with saturated ammonia in absolute ethanol following acidolysis, eliminating the need for ammonium carbonate and improving the isolation of the amidine intermediate. These cumulative optimizations elevate the total yield to nearly 50%, establishing a new benchmark for the reliable pharmaceutical intermediate supplier seeking to maximize output while minimizing raw material costs.
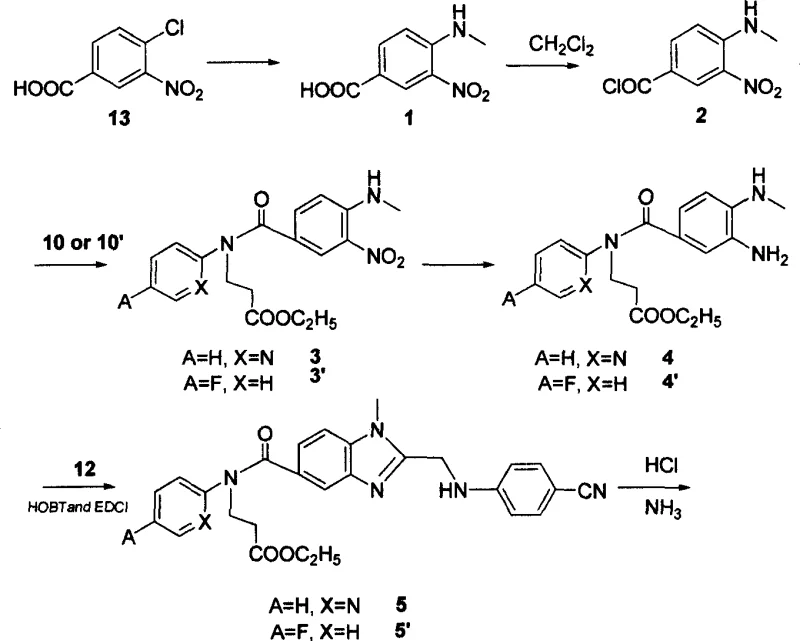
Mechanistic Insights into Benzimidazole Cyclization and Amidination
The mechanistic superiority of this new process is anchored in the strategic selection of coupling reagents and reaction conditions that favor the formation of the 1,2,5-trisubstituted benzimidazole scaffold. In the cyclization step, the reaction between the aniline intermediate (Compound 4) and the cyanoacetic acid derivative (Compound 12) is mediated by HOBT (1-hydroxybenzotriazole) and EDCI (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide). Mechanistically, EDCI activates the carboxylic acid of Compound 12 to form an O-acylisourea intermediate, which is rapidly converted by HOBT into a more stable and less racemization-prone active ester. This active ester then undergoes nucleophilic attack by the ortho-amino group of the aniline, followed by intramolecular condensation to close the imidazole ring. This pathway is significantly more efficient than the CDI-mediated route, as it avoids the formation of stable urea byproducts that can complicate purification. The use of acetic acid reflux in the subsequent step facilitates the dehydration and aromatization of the dihydro-benzimidazole intermediate, ensuring the formation of the fully conjugated heterocyclic system essential for thrombin binding affinity.
Furthermore, the conversion of the nitrile group to the amidine functionality is executed with remarkable precision through a gas-phase protocol. The nitrile intermediate (Compound 5) is first treated with dry hydrogen chloride gas in ethanol to form the imidate hydrochloride salt, a highly reactive electrophile. This is immediately followed by treatment with ammonia gas in ethanol, which displaces the ethoxy group to yield the desired amidine (Compound 6). This gas-phase approach offers distinct advantages over solid-state ammonium carbonate methods, including better stoichiometric control, faster reaction rates, and simplified workup procedures that involve mere concentration and filtration. From an impurity control perspective, this method minimizes the formation of over-hydrolyzed carboxylic acid byproducts, which are common in aqueous amidination attempts. The rigorous control of reaction parameters, such as maintaining temperatures below 0°C during coupling and utilizing anhydrous conditions for amidination, ensures a high-purity profile that meets the stringent requirements for high-purity antithrombin inhibitors intended for clinical use.
How to Synthesize BIBR-953 Efficiently
The synthesis of BIBR-953 outlined in this patent provides a robust framework for laboratory and pilot-scale production, emphasizing operational simplicity and high throughput. The process begins with the nucleophilic aromatic substitution of 3-nitro-4-chlorobenzoic acid with aqueous methylamine to install the aminomethyl group, followed by activation with thionyl chloride. The subsequent coupling and reduction steps are designed to be telescoped where possible, minimizing the number of isolation points and maximizing material throughput. The critical cyclization and amidination steps utilize standard reagents available in any fine chemical facility, removing the dependency on specialized or exotic catalysts. For detailed operational parameters, including specific molar ratios, temperature gradients, and purification techniques such as silica gel column chromatography conditions, operators should refer to the standardized protocols derived from the patent examples.
- Prepare the key intermediate by reacting 3-nitro-4-chlorobenzoic acid with methylamine, followed by conversion to the acyl chloride using thionyl chloride.
- Couple the acyl chloride with ethyl 3-(pyridin-2-ylimino)propanoate, reduce the nitro group, and cyclize with 1-(4-cyanophenylimino)acetic acid using HOBT and EDCI.
- Perform gas-phase amidination using HCl and ammonia, followed by hydrolysis to obtain the final BIBR-953 active pharmaceutical ingredient.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this optimized synthesis route translates into tangible strategic benefits that extend beyond mere technical feasibility. The shift to 3-nitro-4-chlorobenzoic acid as the starting material leverages a commodity chemical that is widely available in the global market, thereby mitigating the risk of supply disruptions associated with custom-synthesized specialty intermediates. The substantial increase in overall yield from 11.1% to nearly 50% implies a drastic reduction in the volume of raw materials required per kilogram of final product, directly impacting the cost of goods sold (COGS). Moreover, the replacement of high-boiling solvents like DMF with volatile solvents like dichloromethane and ethanol facilitates faster drying times and lower energy consumption during solvent recovery, contributing to significant operational expenditure savings. These factors collectively enhance the economic viability of producing BIBR-953, making it a more attractive candidate for generic development or portfolio expansion.
- Cost Reduction in Manufacturing: The elimination of expensive starting materials and the implementation of high-yield coupling reactions fundamentally alter the cost structure of BIBR-953 production. By avoiding the use of CDI and replacing it with the more cost-effective HOBT/EDCI system, the process reduces reagent costs while simultaneously improving yield. The simplified workup procedures, which rely on filtration and concentration rather than complex extractions or chromatographic separations at every step, further reduce labor and consumable costs. This holistic approach to process optimization ensures that the manufacturing of high-purity pharmaceutical intermediates remains economically sustainable even under fluctuating raw material price conditions.
- Enhanced Supply Chain Reliability: Sourcing reliability is paramount for continuous API production, and this route excels by utilizing building blocks that are either commodity chemicals or easily synthesized from them. The starting material, 3-nitro-4-chlorobenzoic acid, is produced by multiple suppliers globally, preventing single-source bottlenecks. Additionally, the robustness of the reaction conditions, which tolerate minor variations in temperature and stoichiometry without significant yield loss, ensures consistent batch-to-bquality. This stability is crucial for reducing lead time for high-purity pharmaceutical intermediates, as it minimizes the need for re-processing or batch rejection, thereby securing a steady flow of material to downstream formulation units.
- Scalability and Environmental Compliance: The process is inherently designed for scalability, with unit operations such as hydrogenation, reflux, and gas treatment being standard in modern multipurpose chemical plants. The reduction in hazardous waste generation, achieved by avoiding DMF and minimizing aqueous washes, aligns with increasingly stringent environmental regulations. The use of ethanol and dichloromethane allows for efficient solvent recycling loops, further enhancing the green chemistry profile of the synthesis. This environmental compatibility not only reduces disposal costs but also future-proofs the manufacturing site against evolving regulatory landscapes, ensuring long-term operational continuity for the production of complex heterocyclic compounds.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis of BIBR-953 and BIBR-1048, based on the detailed disclosures within the patent literature. These insights are intended to clarify the practical implications of the new methodology for stakeholders involved in process development and sourcing. Understanding these nuances is essential for evaluating the feasibility of technology transfer and the potential for immediate implementation in existing manufacturing facilities.
Q: What is the primary advantage of the new BIBR-953 synthesis route?
A: The new route utilizes inexpensive 3-nitro-4-chlorobenzoic acid as a starting material and achieves a total yield of nearly 50%, significantly higher than the 11.1% yield of conventional methods.
Q: How does the new process improve environmental compliance?
A: By replacing DMF with dichloromethane for acyl chloride formation and utilizing efficient coupling agents like HOBT/EDCI, the process simplifies workup procedures and reduces hazardous waste generation.
Q: Is this synthesis method suitable for large-scale production?
A: Yes, the method features simple operation steps, high yields at each stage, and uses readily available reagents, making it highly scalable for commercial manufacturing of antithrombin inhibitors.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable BIBR-953 Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient synthesis routes in the competitive landscape of cardiovascular drug manufacturing. Our team of expert chemists has thoroughly analyzed the methodology disclosed in CN1861596A and is fully equipped to execute this optimized pathway with precision and scale. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with unwavering consistency. Our state-of-the-art facilities are outfitted with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of BIBR-953 or BIBR-1048 meets the highest international standards for pharmaceutical intermediates.
We invite you to collaborate with us to leverage this advanced synthesis technology for your supply chain. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out today to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions that drive value and efficiency in your anticoagulant drug development pipeline.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →