Optimizing Dabigatran Etexilate Production: A Technical Analysis of High-Selectivity Convergent Synthesis
The pharmaceutical industry continuously seeks robust manufacturing pathways for critical anticoagulant therapies, and the synthesis of dabigatran etexilate represents a pinnacle of modern process chemistry challenges. Patent CN102612517A introduces a transformative approach to producing this vital active pharmaceutical ingredient (API) and its analogues, specifically addressing the inefficiencies inherent in earlier generations of synthetic methodology. By shifting away from the cumbersome nitrile hydrolysis routes previously documented in international patents like WO 98/37075, this invention establishes a convergent strategy that leverages protected amidine intermediates and phase-transfer catalysis. The core innovation lies in the ability to couple complex fragments with exceptional regioselectivity, exceeding 99.7%, thereby drastically reducing the burden of downstream purification. This technical breakthrough not only streamlines the molecular construction of the benzimidazole-amidine scaffold but also aligns perfectly with the rigorous demands of green chemistry and industrial scalability required by top-tier generic and innovator drug manufacturers.
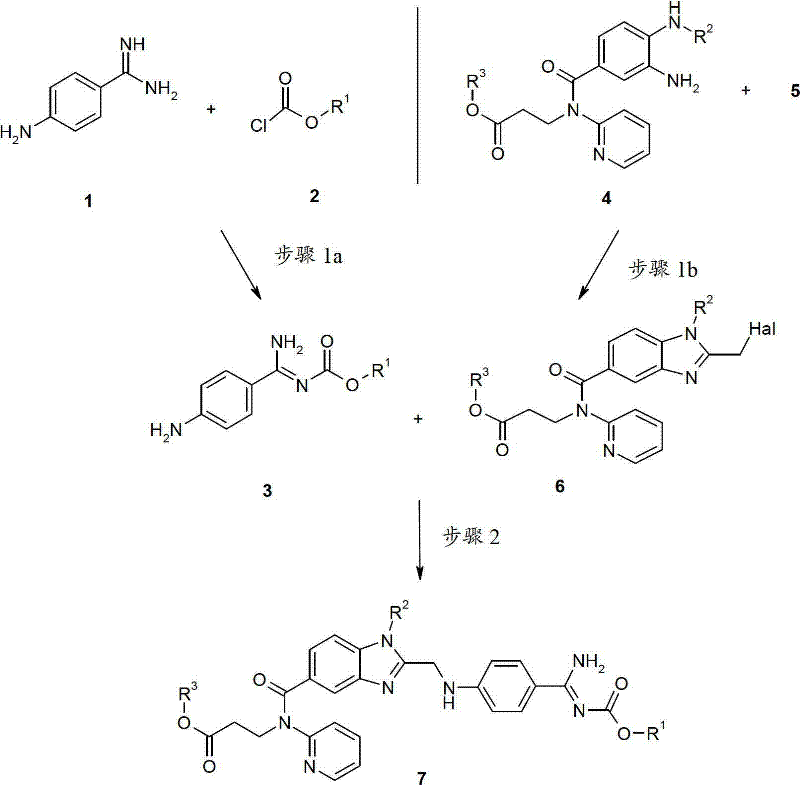
For procurement specialists and supply chain directors, the implications of adopting this methodology are profound, as it replaces hazardous and waste-intensive unit operations with cleaner, higher-yielding transformations. The patent details a multi-step sequence where key intermediates, specifically the protected p-aminobenzamidine derivative (Compound 3) and the functionalized benzimidazole precursor (Compound 6), are synthesized in parallel before being united in a final coupling step. This modular approach allows for greater flexibility in inventory management and risk mitigation, as bottlenecks in one branch of the synthesis do not necessarily halt the entire production line. Furthermore, the reliance on commodity chemicals such as alkyl chloroformates and halogenated acetic acid derivatives ensures that raw material sourcing remains stable and cost-effective, shielding the supply chain from the volatility often associated with exotic reagents. The result is a manufacturing process that delivers high-purity dabigatran etexilate intermediates while simultaneously optimizing the total cost of ownership through reduced waste disposal and simplified operational protocols.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of substituted (4-benzimidazolyl-2-ylmethylamino)-phenyl amidines, such as dabigatran etexilate, relied heavily on the reaction of substituted cyanobenzenes with ammonia to generate the requisite amidine functionality. As highlighted in the background of patent CN102612517A, this traditional pathway, exemplified by WO 98/37075, presents severe drawbacks from a manufacturing perspective. The conversion of nitriles to amidines typically necessitates harsh reaction conditions and generates substantial quantities of acidic waste that require complex and expensive disposal procedures. Moreover, achieving the necessary selectivity often demanded the use of doubly protected amidine species, which added unnecessary synthetic steps, increased material costs, and lowered the overall atom economy of the process. These inefficiencies translated directly into higher production costs and longer lead times, creating significant friction for suppliers attempting to meet the high-volume demands of the global cardiovascular market. The environmental footprint of such processes, characterized by heavy acid usage and difficult workups, also posed compliance challenges in increasingly regulated jurisdictions.
The Novel Approach
In stark contrast, the novel approach disclosed in CN102612517A circumvents these historical bottlenecks by employing a strategically protected mono-amidine coupling strategy. Instead of struggling with the reactivity of free amidines or the harshness of nitrile hydrolysis, the inventors utilize a p-aminobenzamidine carbamate ester (Compound 3) that retains sufficient nucleophilicity for coupling while remaining stable under the reaction conditions. This intermediate is then coupled with a halomethyl-benzimidazole derivative (Compound 6) using a sophisticated phase-transfer catalytic system. The brilliance of this design is its ability to achieve high regioselectivity without the need for cumbersome double-protection groups, effectively collapsing multiple synthetic operations into a single, efficient transformation. By utilizing mild bases like sodium hydroxide or potassium carbonate in biphasic solvent systems, the process avoids the generation of excessive acidic waste, thereby simplifying the effluent treatment profile. This shift represents a paradigm change in how complex anticoagulant intermediates are constructed, prioritizing selectivity and operational simplicity over brute-force chemical transformations.
Mechanistic Insights into Iodide-Catalyzed Amidine Coupling
The heart of this synthetic advancement lies in Step 2, where the coupling of the amidine fragment and the benzimidazole fragment occurs with remarkable precision. The mechanism relies on the activation of the alkyl halide moiety on the benzimidazole intermediate (Compound 6) by iodide ions, typically introduced via potassium iodide or tetrabutylammonium iodide. In this biphasic system, the phase-transfer catalyst facilitates the transport of the deprotonated amidine anion from the aqueous phase into the organic phase, where the electrophilic substitution takes place. The presence of iodide enhances the leaving group ability of the chloride or bromide on the benzimidazole methyl group, forming a more reactive iodomethyl intermediate in situ. This activation lowers the energy barrier for the nucleophilic attack by the amidine nitrogen, allowing the reaction to proceed rapidly at moderate temperatures ranging from 30°C to 60°C. The use of solvents with opposing polarities, such as a mixture of butyl acetate and cyclohexane with water, creates an interfacial environment that maximizes the collision frequency between reactants while maintaining distinct phases for easy separation post-reaction.
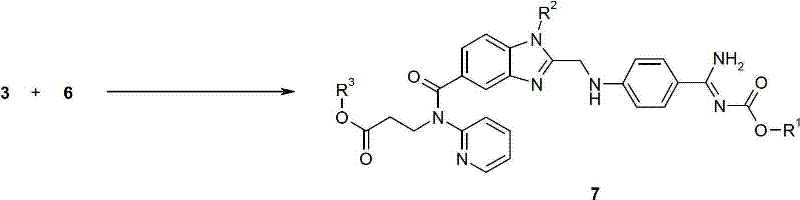
Furthermore, the mechanistic pathway inherently suppresses the formation of regioisomers and over-alkylation byproducts, which are common pitfalls in amidine chemistry. The specific choice of a mono-protected amidine ensures that only one nitrogen atom is available for nucleophilic attack, directing the reaction exclusively towards the desired N-alkylation product. This intrinsic selectivity is further bolstered by the controlled addition of reagents and the maintenance of specific pH levels using weak bases like sodium bicarbonate. From an impurity control perspective, this mechanism is superior because it minimizes the generation of structurally similar byproducts that are difficult to separate via crystallization. The result is a crude product profile that is exceptionally clean, often requiring only a simple wash with solvents like MTBE or butyl acetate to achieve pharmaceutical-grade purity. For R&D directors, understanding this mechanistic nuance is critical, as it validates the robustness of the process against minor fluctuations in reaction parameters, ensuring consistent quality batch after batch.
How to Synthesize Dabigatran Etexilate Intermediates Efficiently
The execution of this synthesis requires precise control over reaction parameters to maximize the benefits of the novel catalytic system. The process begins with the parallel preparation of the two key building blocks: the protected amidine ester and the halomethyl-benzimidazole. Once these intermediates are secured, the critical coupling step is initiated in a reactor equipped for biphasic mixing. The reaction mixture, containing the intermediates, phase-transfer catalyst, and iodide source, is heated to promote the substitution reaction while maintaining vigorous stirring to ensure efficient mass transfer between the organic and aqueous layers. Following the reaction completion, the phases are separated, and the organic layer is subjected to a distillation and solvent swap to induce crystallization of the final product.
- Preparation of Intermediate 3: React p-Aminobenzamidine with alkyl chloroformate in a polar solvent like acetone under basic conditions to form the protected amidine ester.
- Preparation of Intermediate 6: Cyclize the diamine precursor with a halogenated acetic acid derivative (e.g., chloroacetic anhydride) to form the benzimidazole core with a reactive halomethyl group.
- Coupling Reaction (Step 2): React Intermediate 3 and Intermediate 6 in a biphasic solvent system using a phase-transfer catalyst and iodide source to achieve high regioselectivity.
Commercial Advantages for Procurement and Supply Chain Teams
For stakeholders focused on the bottom line and supply continuity, the adoption of the methodology described in CN102612517A offers compelling economic and logistical advantages. The most immediate benefit is the drastic simplification of the waste management profile; by eliminating the need for large-scale acid neutralization associated with nitrile hydrolysis, manufacturers can significantly reduce their environmental compliance costs and hazardous waste disposal fees. This reduction in chemical intensity also translates to lower raw material consumption per kilogram of finished product, directly impacting the variable cost of goods sold. Moreover, the use of common, non-proprietary solvents such as butyl acetate, ethyl acetate, and cyclohexane ensures that the supply chain is not vulnerable to shortages of specialized reagents. These solvents are widely available from multiple global suppliers, providing procurement managers with the leverage to negotiate better pricing and secure long-term contracts. The robustness of the process also means that production schedules are less likely to be disrupted by failed batches or complex purification delays, ensuring a steady flow of material to downstream formulation units.
- Cost Reduction in Manufacturing: The elimination of expensive coupling agents and the reduction in purification steps lead to substantial cost savings. By avoiding the use of doubly protected intermediates, the process reduces the number of synthetic steps, which cumulatively lowers labor, energy, and equipment usage costs. The high yields reported in the patent examples, such as 97.2% for Step 1a and 90.3% for Step 1b, indicate a highly efficient use of starting materials, minimizing the financial loss associated with unreacted feedstock. Additionally, the ability to isolate products via simple crystallization rather than column chromatography or complex distillations further drives down processing costs, making the final API more price-competitive in the generic market.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals and standard unit operations enhances the resilience of the supply chain. Since the process does not depend on fragile catalysts or unstable reagents that require cold-chain logistics, inventory management becomes more straightforward and less costly. The parallel synthesis of intermediates allows for the stocking of key precursors, decoupling the production of the final coupled product from potential upstream delays. This modularity ensures that even if one supply line faces temporary disruption, the overall production timeline can be maintained by drawing from existing intermediate inventories, thereby guaranteeing on-time delivery to customers.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, utilizing reaction conditions that are easily replicated in large-scale stainless steel reactors. The moderate temperature ranges and atmospheric pressure operations eliminate the need for specialized high-pressure or cryogenic equipment, reducing capital expenditure requirements for new production lines. From an environmental standpoint, the reduced generation of acidic waste and the use of recyclable solvents align with modern sustainability goals, facilitating easier regulatory approval in strict jurisdictions. This compliance advantage protects the manufacturer from future regulatory shocks and positions the product as a preferred choice for environmentally conscious pharmaceutical partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These answers are derived directly from the experimental data and claims within the patent documentation to provide accurate guidance for technical teams evaluating this technology. Understanding these nuances is essential for assessing the feasibility of technology transfer and the potential for process optimization at a commercial scale.
Q: What is the primary advantage of this synthesis method over conventional routes?
A: The primary advantage is the elimination of harsh nitrile hydrolysis steps required in older methods (like WO 98/37075), which generated large amounts of acid waste. This new route uses a protected amidine coupling that achieves >99.7% regioselectivity without needing double protection groups.
Q: How does the process ensure high purity for pharmaceutical applications?
A: High purity is ensured through the use of specific phase-transfer catalysts (tetrabutylammonium iodide) and iodide activation in Step 2, which minimizes side reactions. Additionally, the crystallization steps using solvents like MTBE and butyl acetate effectively remove impurities.
Q: Is this process scalable for commercial production?
A: Yes, the process utilizes common industrial solvents such as butyl acetate, ethyl acetate, and cyclohexane, and operates at moderate temperatures (30-70°C), making it highly suitable for commercial scale-up without requiring specialized cryogenic or high-pressure equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Dabigatran Etexilate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and proven manufacturing capabilities. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated chemistry described in CN102612517A can be seamlessly translated into high-volume manufacturing. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of dabigatran etexilate intermediate meets the exacting standards of the global pharmaceutical industry. Our commitment to quality is matched only by our dedication to process safety and environmental stewardship, making us an ideal partner for long-term supply agreements.
We invite procurement leaders and R&D directors to engage with us for a Customized Cost-Saving Analysis tailored to your specific production needs. By leveraging our optimized version of this convergent synthesis, we can help you achieve significant reductions in COGS while maintaining the highest levels of product integrity. We encourage you to contact our technical procurement team to request specific COA data and route feasibility assessments, allowing you to make informed decisions that will strengthen your supply chain and enhance your market competitiveness. Let us collaborate to bring this advanced therapeutic to patients more efficiently and economically.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →