Scalable Production of High-Purity Prulifloxacin Intermediates via Novel Bromomethyl Route
Scalable Production of High-Purity Prulifloxacin Intermediates via Novel Bromomethyl Route
The pharmaceutical industry continuously seeks robust, high-yield pathways for the production of critical antibiotic intermediates, particularly for fourth-generation fluoroquinolones like Prulifloxacin. A pivotal advancement in this domain is detailed in patent CN1583757A, which discloses a revolutionary synthesis method that fundamentally alters the economic and technical landscape of producing this vital active pharmaceutical ingredient (API). Unlike traditional multi-step sequences that suffer from cumulative yield losses and purification bottlenecks, this novel methodology leverages a strategic coupling of a specialized bromomethyl-dioxolone derivative with a fluorinated quinoline carboxylic acid core. For R&D directors and procurement strategists evaluating reliable API intermediate suppliers, understanding the mechanistic elegance and operational simplicity of this route is paramount. The patent data reveals a process that not only simplifies the reaction sequence but also dramatically enhances the purity profile of the crude product, achieving levels greater than 97.5% prior to final recrystallization. This technical breakthrough translates directly into reduced manufacturing costs and enhanced supply chain reliability for global pharmaceutical manufacturers seeking to optimize their production of broad-spectrum antibacterial agents.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Prulifloxacin and its precursors has been plagued by significant technical inefficiencies that hinder cost-effective commercialization. As outlined in the background art of the patent, conventional routes typically rely on starting materials such as 2,4,5-trifluorobenzoic acid, 3,4-difluoroaniline, or 2-amino-4,5-dihydroxybenzoic acid. While these pathways eventually converge on the formation of the key quinoline intermediate (Formula IV), the subsequent steps to attach the piperazine side chain are notoriously problematic. The traditional approach involves synthesizing the ethyl ester of the quinoline acid, followed by a condensation reaction with piperazine, and finally, a hydrolysis step to obtain the free acid (Formula V). This sequence is not only laden with trivial and tedious operational steps but also suffers from inherently low yields, often hovering around merely 40% when calculated from the quinoline intermediate. Furthermore, the instability of the resulting acid salts and the complexity of the crystallization aftertreatment make it exceedingly difficult to obtain products of higher degrees of purity. These factors collectively render the conventional technology unstable and poorly suited for suitability for industrialized production, creating substantial bottlenecks for supply chain heads managing large-volume commitments.
The Novel Approach
In stark contrast to the convoluted legacy methods, the novel approach presented in patent CN1583757A introduces a streamlined strategy that bypasses the inefficient ester-hydrolysis sequence entirely. The innovation centers on the use of 4-bromomethyl-5-methyl-1,3-dioxacycloene pentadione (Formula VI) as a highly reactive electrophilic starting material. 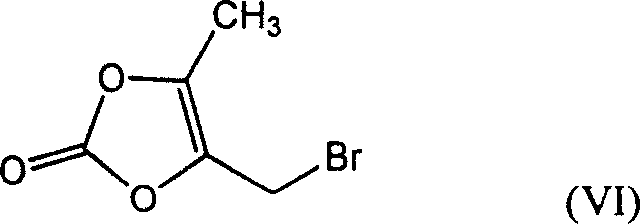 This specialized reagent undergoes a direct addition reaction with piperazine to generate the key intermediate, 5-methyl-4-piperazinomethyl-1,3-dioxacycloenepentenedione (Formula II), with exceptional efficiency. This intermediate is then coupled directly with the fluorinated quinoline carboxylic acid (Formula III),
This specialized reagent undergoes a direct addition reaction with piperazine to generate the key intermediate, 5-methyl-4-piperazinomethyl-1,3-dioxacycloenepentenedione (Formula II), with exceptional efficiency. This intermediate is then coupled directly with the fluorinated quinoline carboxylic acid (Formula III), 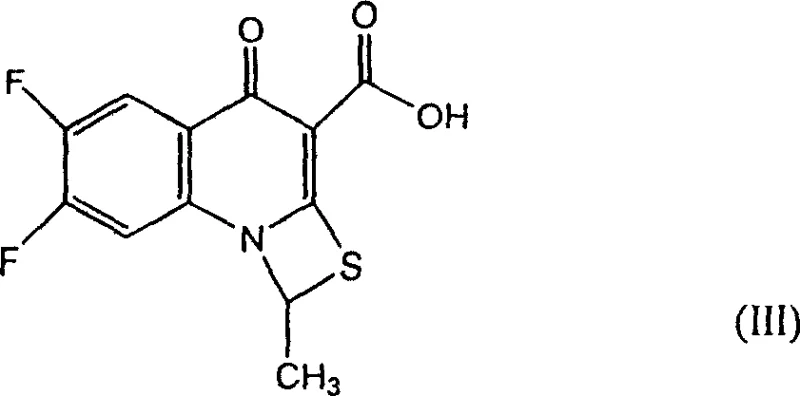 eliminating the need for separate esterification and hydrolysis steps. By restructuring the synthetic logic to prioritize direct nucleophilic substitution on the side chain precursor, the new method achieves a total product yield of more than 58%, representing a massive 45% increase compared to the prior art. This shift not only simplifies the process flow but also ensures that the crude product emerges with a purity exceeding 97.5%, drastically reducing the load on downstream purification units and offering a clear pathway for cost reduction in antibiotic manufacturing.
eliminating the need for separate esterification and hydrolysis steps. By restructuring the synthetic logic to prioritize direct nucleophilic substitution on the side chain precursor, the new method achieves a total product yield of more than 58%, representing a massive 45% increase compared to the prior art. This shift not only simplifies the process flow but also ensures that the crude product emerges with a purity exceeding 97.5%, drastically reducing the load on downstream purification units and offering a clear pathway for cost reduction in antibiotic manufacturing.
Mechanistic Insights into Nucleophilic Substitution and Coupling
The core chemical transformation driving this improved synthesis is a classic yet optimized nucleophilic substitution reaction, specifically an SN2 mechanism, facilitated by the high reactivity of the bromomethyl group in Formula VI. In the first stage, the secondary amine nitrogen of the piperazine ring acts as a potent nucleophile, attacking the methylene carbon attached to the bromine atom in the dioxolone derivative. This reaction is preferably carried out in polar aprotic solvents such as N,N-dimethylformamide (DMF), which stabilizes the transition state and enhances the nucleophilicity of the piperazine without solvating it too strongly. The displacement of the bromide ion is rapid and clean, leading to the formation of the C-N bond that links the piperazine ring to the dioxolone moiety. The resulting intermediate (Formula II) is chemically robust, allowing for straightforward isolation via organic solvent extraction using chloroform and subsequent recrystallization in ethyl acetate-petroleum ether mixtures. This mechanistic clarity ensures that impurities related to over-alkylation or incomplete reaction are minimized, providing a high-quality building block for the final coupling step.
The second critical mechanistic phase involves the coupling of the newly formed piperazine intermediate (Formula II) with the quinoline carboxylic acid (Formula III). This step requires careful control of reaction conditions to ensure the selective alkylation of the distal nitrogen of the piperazine ring while preserving the integrity of the sensitive fluorinated quinoline core. The reaction is conducted in DMF under basic conditions, often utilizing alkali metal carbonates or bicarbonates to deprotonate the piperazine nitrogen, thereby increasing its nucleophilicity for the attack on the electrophilic center (though in this specific patent embodiment, the coupling logic implies the pre-formed intermediate reacts with the acid, potentially via an activated species or direct thermal coupling depending on specific activation not fully detailed but implied by the high yield). The result is the formation of the final Prulifloxacin structure with the correct stereochemistry and substitution pattern. The high crude purity (>97.5%) indicates that side reactions, such as defluorination of the quinoline ring or degradation of the dioxolone ring, are effectively suppressed. This level of impurity control is vital for meeting the stringent regulatory standards required for high-purity pharmaceutical intermediates, ensuring that the final API meets all pharmacopoeial specifications with minimal rework.
How to Synthesize Prulifloxacin Efficiently
The synthesis of Prulifloxacin via this novel route offers a distinct operational advantage by consolidating multiple transformation steps into a more direct sequence. The process begins with the preparation of the key side-chain intermediate through the reaction of 4-bromomethyl-5-methyl-1,3-dioxacycloene pentadione with anhydrous piperazine in DMF at room temperature, followed by a straightforward workup involving water precipitation and chloroform extraction. Once the intermediate (Formula II) is secured as a red solid, it is reacted with the quinoline acid (Formula III) in the presence of a base like potassium bicarbonate in DMF at moderate temperatures (around 50°C). The reaction progress is easily monitored via thin-layer chromatography (TLC), and upon completion, the product precipitates upon the addition of ice water, allowing for simple filtration.
- React 4-bromomethyl-5-methyl-1,3-dioxacycloene pentadione with piperazine in DMF to form the key piperazine-methyl intermediate.
- Purify the intermediate via chloroform extraction and recrystallization in ethyl acetate-petroleum ether.
- Couple the purified intermediate with 6,7-difluoro-1-methyl-4-oxo-4H-[1,3]thiazino[3,2-a]quinoline-3-carboxylic acid in DMF under basic conditions.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthesis route presents a compelling value proposition centered on operational efficiency and cost predictability. The primary driver of value is the significant simplification of the process workflow, which directly correlates to reduced operational expenditures (OPEX). By eliminating the need for separate esterification and hydrolysis steps, the facility saves on solvent consumption, energy usage for heating and cooling cycles, and labor hours associated with additional unit operations. Furthermore, the dramatic improvement in crude purity means that the burden on the purification department is substantially lightened. Less solvent is required for recrystallization, and the loss of product during mother liquor handling is minimized. This efficiency gain allows for a higher throughput of material within the same existing infrastructure, effectively increasing capacity without capital investment. Additionally, the use of commodity chemicals like DMF, chloroform, and acetonitrile ensures that raw material sourcing remains stable and insulated from the volatility often associated with exotic reagents or specialized catalysts.
- Cost Reduction in Manufacturing: The elimination of transition steps such as ester hydrolysis removes entire categories of cost, including the reagents and solvents required for those specific transformations. The high yield of the final step (over 73%) and the overall yield increase to >58% mean that less starting material is wasted per kilogram of final product. This material efficiency is a direct contributor to margin expansion, as the cost of goods sold (COGS) is lowered through better atom economy and reduced waste disposal costs. Moreover, the stability of the intermediates reduces the risk of batch failures, which are a hidden but significant cost in pharmaceutical manufacturing.
- Enhanced Supply Chain Reliability: The reliance on commercially available and industrially mature starting materials, such as the bromomethyl-dioxolone derivative and the quinoline acid (which can be sourced from established suppliers or produced via known methods like JP85-73885), mitigates supply risk. The process does not depend on single-source specialty catalysts or air-sensitive reagents that could disrupt production schedules. The robustness of the reaction conditions—operating at moderate temperatures and atmospheric pressure—further ensures that the process can be run consistently across different manufacturing sites, providing supply chain heads with the confidence of continuity and the ability to dual-source if necessary.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, the simplified workflow results in a smaller environmental footprint. Fewer reaction steps equate to less solvent waste and lower energy consumption, aligning with modern green chemistry principles. The ability to isolate the product via simple precipitation and filtration, rather than complex chromatographic separations, makes the process highly scalable from pilot plant to commercial tonnage. The high purity of the crude product also minimizes the generation of hazardous waste streams associated with aggressive purification techniques, facilitating easier compliance with increasingly stringent environmental regulations governing pharmaceutical effluent.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent literature, offering a transparent view of the process capabilities. Understanding these details is crucial for technical teams evaluating the feasibility of technology transfer or scale-up initiatives.
Q: What is the primary advantage of the novel synthesis route for Prulifloxacin?
A: The novel route utilizing 4-bromomethyl-5-methyl-1,3-dioxacycloene pentadione eliminates the need for complex ester hydrolysis and condensation steps found in conventional methods, resulting in a total yield increase from approximately 40% to over 58% and crude purity exceeding 97.5%.
Q: How does this process address the stability issues of Prulifloxacin intermediates?
A: Conventional methods often struggle with unstable acid salts and difficult crystallization. This new approach generates a stable off-white solid crude product that can be easily purified via simple acetonitrile recrystallization, significantly improving process stability and industrial suitability.
Q: Is this synthesis method suitable for large-scale commercial production?
A: Yes, the process uses common industrial solvents like DMF and acetonitrile and avoids exotic catalysts. The high crude purity (>97.5%) reduces the burden on downstream purification, making it highly viable for metric-ton scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Prulifloxacin Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and proven manufacturing capabilities. The synthesis route described in patent CN1583757A exemplifies the type of process optimization our CDMO team excels at executing. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of high yield and purity are realized in actual manufacturing batches. Our facilities are equipped with rigorous QC labs and advanced analytical instrumentation to verify that every batch of Prulifloxacin intermediate meets stringent purity specifications, adhering to the highest international standards for pharmaceutical quality. We understand that consistency is key for your regulatory filings and market supply, and our quality management systems are designed to deliver exactly that.
We invite you to engage with our technical procurement team to discuss how this optimized synthesis route can be integrated into your supply chain. Whether you require a Customized Cost-Saving Analysis to quantify the potential economic benefits for your specific operation or need specific COA data to validate the quality profile, our experts are ready to assist. We encourage you to request route feasibility assessments to determine the best path forward for your project. By leveraging our manufacturing prowess and your market vision, we can jointly unlock the full commercial potential of this advanced antibiotic intermediate.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →