Scalable Production of Carfilzomib Using Novel Phenylketene-Mediated Coupling Technology
The pharmaceutical industry continuously seeks robust synthetic pathways for complex oncology therapeutics, and patent CN112830957A presents a breakthrough methodology for the efficient preparation of Carfilzomib, a second-generation proteasome inhibitor approved for treating refractory multiple myeloma. This novel approach addresses critical bottlenecks in peptide synthesis by utilizing phenylketene as a specialized condensing agent, fundamentally altering the activation landscape of chiral carboxylic acids. Unlike traditional coupling reagents that often induce racemization or require harsh conditions, this technology enables a stepwise assembly of the pentapeptide-like structure with exceptional stereochemical fidelity. The process initiates with readily available chloroacetic acid and proceeds through a series of activation and condensation cycles, ultimately delivering the target molecule with a remarkable total yield ranging from 68 to 72 percent. For R&D directors and procurement managers alike, this represents a significant leap forward in process chemistry, offering a reliable API intermediate supplier pathway that balances high purity with operational simplicity.
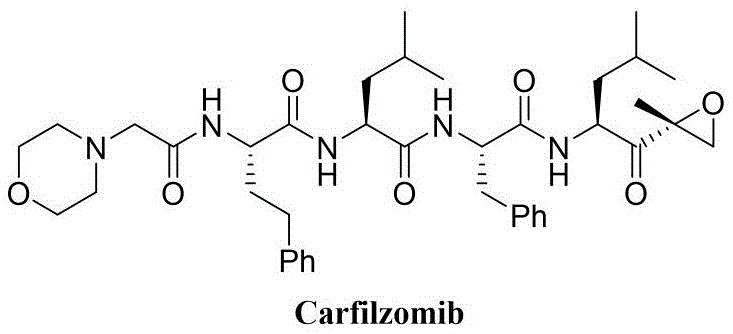
Carfilzomib possesses a complex architecture featuring multiple amide linkages and a critical epoxy ketone pharmacophore, necessitating a synthesis strategy that preserves chirality at every alpha-carbon. The background art reveals that existing methods, while functional, often struggle with the activation of long-chain polypeptide carboxylic acids, leading to serious racemization issues and diminished overall yields typically below 45 percent. Furthermore, conventional strategies frequently involve expensive reagents and complex purification protocols that hinder industrial scalability. The invention described in CN112830957A overcomes these limitations by introducing a ketene-mediated activation protocol that is both atom-economical and gentle on sensitive functional groups. By shifting the paradigm from standard carbodiimide or phosphonium-based coupling to phenylketene activation, the process eliminates the formation of difficult-to-remove byproducts and ensures that the optical purity of the final drug substance meets stringent regulatory specifications without the need for extensive chiral chromatography.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Carfilzomib has been plagued by the inherent difficulties of assembling long peptide chains while maintaining stereochemical integrity. Conventional methods often rely on activating agents that generate significant amounts of waste or require strict anhydrous conditions that are difficult to maintain on a large scale. A primary drawback identified in prior art, such as various patent applications from the mid-2000s and 2010s, is the tendency for racemization at the alpha-position of the carboxyl group during the activation of long-chain intermediates. This racemization not only reduces the yield of the desired enantiomer but also creates diastereomeric impurities that are notoriously difficult to separate, thereby complicating the downstream purification process. Additionally, the reliance on expensive protecting group strategies and coupling reagents drives up the cost of goods significantly, making the final API less accessible. The cumulative effect of these inefficiencies is a total yield that rarely exceeds 45 percent, which is economically unsustainable for high-volume commercial production of life-saving oncology medications.
The Novel Approach
The innovative strategy outlined in the patent utilizes phenylketene as a powerful yet mild condensing agent to drive the formation of amide bonds with high efficiency. This approach allows for a modular, stepwise construction of the Carfilzomib backbone, starting from chloroacetic acid and sequentially adding homophenylalanine, leucine, phenylalanine, and finally the epoxy ketone amino acid derivative. The use of phenylketene ensures that the activation of the carboxylic acid occurs without compromising the chiral center, effectively solving the racemization problem that plagues traditional methods. As illustrated in the reaction scheme below, the process flows logically through activated esters (E1, E2, E3, E4) which are then coupled with the respective amino acid components. This methodology not only simplifies the operational workflow by allowing for one-pot deprotection and activation sequences but also drastically improves the atom economy of the reaction. The result is a streamlined process that delivers high-purity intermediates and a final product with a total yield of 68-72 percent, representing a substantial improvement over the state of the art.
Mechanistic Insights into Phenylketene-Mediated Amide Bond Formation
The core mechanistic advantage of this synthesis lies in the unique reactivity of phenylketene towards carboxylic acids. When chloroacetic acid or the growing peptide chain reacts with phenylketene in a solvent like 1,2-Dichloroethane (DCE), it forms a highly reactive mixed anhydride or activated ester species. This activated intermediate is sufficiently electrophilic to react rapidly with the amine group of the incoming amino acid, even under mild conditions, yet it is stable enough to prevent the abstraction of the alpha-proton that leads to racemization. This delicate balance is crucial for preserving the biological activity of the final proteasome inhibitor, as even minor epimerization can render the molecule inactive or toxic. The reaction conditions are typically maintained between 20°C and 60°C, avoiding the thermal stress that can degrade sensitive peptide bonds or the epoxy ketone warhead. Furthermore, the byproduct of this activation is minimal and easily removable, contributing to the high purity of the crude product before any chromatographic purification is attempted.
In the final stages of the synthesis, the chloroacetyl-terminated peptide intermediate undergoes a nucleophilic substitution with morpholine to install the N-terminal cap. This specific transformation is catalyzed by potassium iodide (KI) in Tetrahydrofuran (THF), which enhances the nucleophilicity of the morpholine and facilitates the displacement of the chloride ion. The use of a catalytic amount of KI is a strategic choice that accelerates the reaction rate without introducing stoichiometric amounts of heavy metals, aligning with green chemistry principles. The entire sequence is designed to minimize the number of isolation steps; for instance, the patent describes 'two-step one-pot' and 'three-step one-pot' variations where deprotection, activation, and coupling can occur in sequence without isolating the unstable free acid intermediates. This telescoping of steps not only saves time but also reduces solvent consumption and material loss, which are critical factors when evaluating the commercial viability of complex peptide mimetics manufacturing.
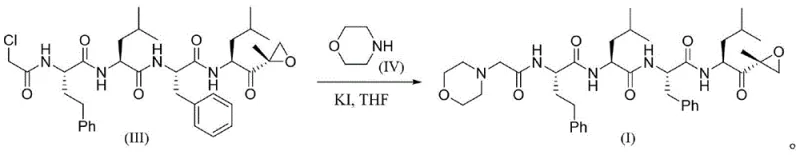
How to Synthesize Carfilzomib Efficiently
The synthesis of Carfilzomib via this novel route involves a logical progression of coupling reactions that build the molecule from the N-terminus towards the C-terminus warhead. The process begins with the activation of chloroacetic acid, followed by sequential additions of protected amino acids, with deprotection and re-activation occurring between each coupling event. The detailed standardized synthesis steps, including specific molar ratios, solvent volumes, and reaction times for each stage from the formation of Activated Ester E1 to the final morpholine substitution, are provided in the structured guide below for technical reference.
- Activate chloroacetic acid with phenylketene in DCE to form activated ester E1, then couple with protected homophenylalanine.
- Deprotect the carboxyl group of the dipeptide, react with phenylketene again, and couple with protected leucine to extend the chain.
- Repeat the deprotection and phenylketene activation cycle to couple protected phenylalanine, forming the tripeptide intermediate.
- Perform final deprotection and activation, then couple with the epoxy ketone amino acid derivative to form the full backbone.
- React the chloroacetyl intermediate with morpholine in THF using potassium iodide catalyst to yield final Carfilzomib.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this phenylketene-mediated synthesis offers tangible benefits that extend beyond mere chemical elegance. The primary advantage is the drastic simplification of the supply chain for raw materials; chloroacetic acid and standard protected amino acids are commodity chemicals available from multiple global sources, reducing the risk of single-supplier dependency. Moreover, the elimination of exotic or prohibitively expensive coupling reagents translates directly into cost reduction in pharmaceutical manufacturing. The high yield of 68-72 percent means that less starting material is required to produce the same amount of API, effectively lowering the cost per kilogram of the final product. Additionally, the mild reaction conditions reduce the energy load on the manufacturing facility, as there is no need for cryogenic cooling or extreme heating, further contributing to operational expenditure savings.
- Cost Reduction in Manufacturing: The economic impact of this process is driven by the high atom economy and the avoidance of costly chiral resolving agents. Since the method prevents racemization inherently, there is no need for expensive chiral HPLC separation steps to remove unwanted enantiomers, which are typically a major cost driver in peptide synthesis. The use of common solvents like DCE, DMF, and THF allows for efficient solvent recovery and recycling systems to be implemented, minimizing waste disposal costs. Furthermore, the high yield reduces the volume of waste generated per unit of product, aligning with environmental compliance goals and reducing the financial burden of waste treatment.
- Enhanced Supply Chain Reliability: By relying on robust and widely available starting materials, manufacturers can secure a more stable supply chain that is less susceptible to market fluctuations. The simplicity of the operation, which does not require specialized equipment for handling air-sensitive reagents or extreme temperatures, means that the process can be transferred to a wider range of CDMO partners globally. This flexibility ensures continuity of supply, which is critical for meeting the demands of the oncology market. The reduced complexity of the workup procedures also shortens the batch cycle time, allowing for faster turnover and reducing the lead time for high-purity oncology APIs.
- Scalability and Environmental Compliance: The process is inherently scalable due to its reliance on liquid-phase reactions with manageable exotherms. The absence of heavy metal catalysts in the main coupling steps simplifies the purification process and ensures that the final product meets strict limits for residual metals without requiring additional scavenging steps. This feature is particularly important for regulatory approval and patient safety. The overall green profile of the synthesis, characterized by high yields and reduced solvent usage per unit of product, supports sustainability initiatives and helps companies meet increasingly stringent environmental regulations regarding volatile organic compound (VOC) emissions and chemical waste.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and technical specifications provided in the patent documentation, ensuring accuracy and relevance for decision-makers evaluating this pathway for potential licensing or contract manufacturing.
Q: How does the phenylketene method improve yield compared to traditional coupling?
A: Traditional methods often suffer from racemization and low yields (below 45%) during long-chain peptide activation. The phenylketene method operates under mild conditions that prevent racemization at chiral centers, achieving a total yield of 68-72%.
Q: What are the key solvents used in this scalable synthesis route?
A: The process utilizes common industrial solvents including 1,2-Dichloroethane (DCE) for activation steps, N,N-Dimethylformamide (DMF) for coupling reactions, and Tetrahydrofuran (THF) for the final morpholine substitution, ensuring ease of handling and recovery.
Q: Is this method suitable for large-scale commercial production?
A: Yes, the method features simple operations, mild reaction temperatures (often room temperature to 35°C), and avoids expensive transition metal catalysts in the main coupling steps, making it highly suitable for commercial scale-up of complex peptide mimetics.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Carfilzomib Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this phenylketene-mediated synthesis route for the commercial production of Carfilzomib. As a leading CDMO expert, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this patent are fully realized in a GMP environment. Our rigorous QC labs and stringent purity specifications guarantee that every batch of Carfilzomib intermediate or API produced meets the highest international standards, providing our partners with the confidence needed to navigate the complex regulatory landscape of oncology drug development.
We invite you to engage with our technical procurement team to discuss how this innovative synthesis can optimize your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain specific insights into how switching to this high-yield route can impact your bottom line. We encourage potential partners to contact us for specific COA data and route feasibility assessments tailored to your project requirements, ensuring a seamless transition from laboratory scale to industrial manufacturing.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →