Advanced Synthesis of USP7 Inhibitor Intermediates for Commercial Oncology Applications
The pharmaceutical landscape for oncology therapeutics is continuously evolving, with a significant focus on targeting the ubiquitin-proteasome system to regulate protein stability within cancer cells. A pivotal development in this arena is documented in patent CN115215883A, which discloses a novel class of USP7 inhibitors characterized by a unique thieno[3,2-b]pyridine and indole fused scaffold. This intellectual property represents a substantial leap forward in the design of small molecule deubiquitinase inhibitors, offering enhanced selectivity and potent in vivo activity compared to earlier generations of compounds. For R&D directors and procurement specialists, understanding the chemical architecture and synthetic accessibility of these molecules is crucial for integrating them into broader drug discovery pipelines. The patent outlines not only the biological efficacy but also provides robust synthetic methodologies that pave the way for reliable manufacturing. As a reliable USP7 inhibitor supplier, analyzing these structural nuances allows us to anticipate supply chain needs and ensure the availability of high-quality intermediates for downstream API synthesis.
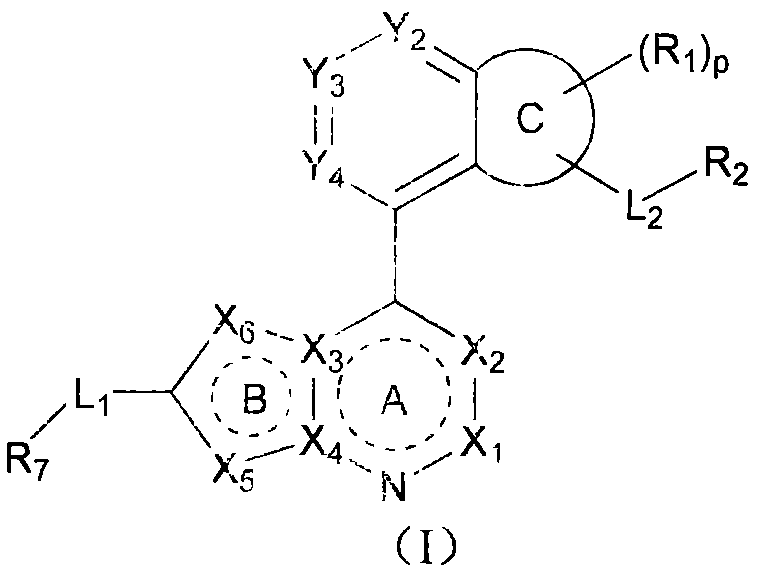
The development of effective cancer therapeutics often hinges on the ability to disrupt specific protein-protein interactions that drive tumor growth. USP7, also known as HAUSP, plays a critical role in stabilizing MDM2, which in turn regulates the tumor suppressor p53. By inhibiting USP7, these novel compounds can restore p53 function, leading to cell cycle arrest and apoptosis in tumor cells. The chemical structures disclosed in the patent feature a diverse array of substituents on the core heterocyclic system, allowing for fine-tuning of pharmacokinetic properties. This structural flexibility is a key advantage for medicinal chemists aiming to optimize solubility and metabolic stability. The ability to access these complex scaffolds through efficient synthetic routes is a major consideration for supply chain heads who must ensure continuity of material for clinical trials. The patent data suggests that these inhibitors exhibit low nanomolar potency, making them highly attractive candidates for further development in treating various malignancies including leukemia and solid tumors.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of complex heterocyclic inhibitors like those targeting USP7 has been plagued by challenges related to regioselectivity and low overall yields. Conventional methods often rely on harsh reaction conditions that can compromise sensitive functional groups, leading to the formation of difficult-to-remove impurities. For instance, traditional cross-coupling reactions might require excessive amounts of expensive palladium catalysts or toxic ligands, which complicates the purification process and increases the environmental footprint. Furthermore, older synthetic routes frequently involve multiple protection and deprotection steps that add significant time and cost to the manufacturing process. These inefficiencies can result in prolonged lead times and higher costs for high-purity USP7 inhibitors, creating bottlenecks for pharmaceutical companies aiming to bring new therapies to market quickly. The lack of scalable processes for earlier generation inhibitors has often limited their availability for extensive preclinical testing.
The Novel Approach
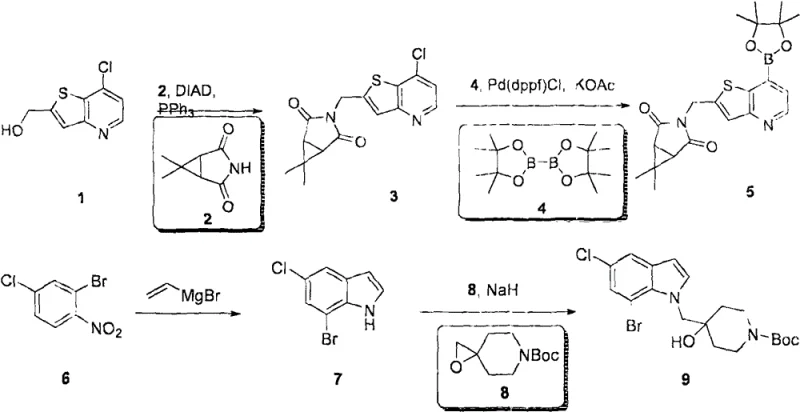
In contrast, the methodology described in CN115215883A introduces a streamlined synthetic strategy that addresses many of these historical pain points. The novel approach utilizes a convergent synthesis design, where key fragments such as the thieno[3,2-b]pyridine and the substituted indole are prepared independently and then coupled together. This strategy significantly improves the overall efficiency and allows for greater flexibility in introducing diverse substituents. The use of mild reaction conditions, such as the specific palladium-catalyzed coupling steps detailed in the examples, ensures that sensitive moieties remain intact throughout the synthesis. This results in a cleaner reaction profile and simplifies the downstream purification, ultimately leading to cost reduction in oncology intermediate manufacturing. By optimizing the sequence of reactions, the patent demonstrates a pathway that is not only chemically elegant but also practically viable for large-scale production, ensuring a stable supply of these critical therapeutic agents.
Mechanistic Insights into USP7 Deubiquitinase Inhibition
The mechanism of action for these compounds involves the specific binding to the catalytic domain of the USP7 enzyme, thereby preventing the removal of ubiquitin chains from substrate proteins. This inhibition disrupts the normal recycling of ubiquitin and leads to the accumulation of ubiquitinated proteins, which triggers cellular stress responses. Structurally, the compounds interact with key residues in the USP7 active site through hydrogen bonding and hydrophobic interactions. The thieno[3,2-b]pyridine core serves as a rigid scaffold that positions the pharmacophoric groups precisely within the enzyme pocket. This precise fit is essential for achieving the high selectivity observed in the biological data, minimizing off-target effects on other deubiquitinases. Understanding this mechanistic interaction is vital for R&D teams as it guides the design of next-generation analogs with improved potency. The patent data indicates that specific substitutions on the indole nitrogen and the piperidine ring significantly influence binding affinity, providing a clear structure-activity relationship for future optimization efforts.
Impurity control is another critical aspect of the mechanistic understanding, particularly regarding the formation of byproducts during the coupling reactions. The synthetic route employs specific reagents and conditions that minimize the formation of regioisomers, which are common impurities in heterocyclic chemistry. For example, the use of specific bases and solvents in the alkylation steps ensures that the reaction proceeds at the desired nitrogen atom rather than competing carbon sites. This level of control is essential for meeting the stringent purity specifications required for pharmaceutical intermediates. The purification methods described, such as flash chromatography with optimized solvent systems, are designed to effectively separate the target compound from closely related impurities. This focus on purity ensures that the final material is suitable for biological testing without interference from synthetic byproducts, thereby providing reliable data for clinical decision-making.
How to Synthesize USP7 Inhibitor Intermediates Efficiently
The synthesis of these complex molecules requires a systematic approach that balances chemical efficiency with operational safety. The patented process outlines a series of well-defined steps that transform readily available starting materials into the final inhibitor scaffold. Key to this process is the management of reaction parameters such as temperature and stoichiometry to maximize yield. The initial steps involve the construction of the core heterocyclic rings, followed by the strategic introduction of side chains that confer biological activity. Detailed standard operating procedures for these transformations are essential for ensuring reproducibility across different manufacturing sites. The following guide summarizes the critical stages involved in producing these high-value intermediates, highlighting the key reagents and conditions necessary for success.
- Initial construction of the thieno[3,2-b]pyridine core via substitution reactions using protected intermediates.
- Palladium-catalyzed cross-coupling to attach the indole moiety, ensuring high regioselectivity and yield.
- Final deprotection and functionalization steps to yield the active USP7 inhibitor intermediate ready for salt formation.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this novel synthetic route offers significant advantages for procurement and supply chain management. The streamlined nature of the synthesis reduces the number of unit operations required, which directly translates to lower manufacturing costs and reduced resource consumption. By eliminating the need for exotic reagents or complex purification techniques, the process becomes more accessible to a wider range of contract manufacturing organizations. This accessibility enhances supply chain reliability by diversifying the potential vendor base and reducing the risk of single-source dependency. Furthermore, the robustness of the chemistry ensures consistent quality output, which is critical for maintaining regulatory compliance throughout the drug development lifecycle. These factors collectively contribute to a more resilient and cost-effective supply chain for oncology therapeutics.
- Cost Reduction in Manufacturing: The synthetic route described avoids the use of prohibitively expensive catalysts and reagents, relying instead on commercially available materials that are cost-effective at scale. The high yields achieved in key coupling steps minimize material waste, further driving down the cost of goods. Additionally, the simplified purification process reduces the consumption of solvents and chromatography media, which are significant cost drivers in pharmaceutical manufacturing. These efficiencies allow for substantial cost savings without compromising the quality of the final product, making the therapy more economically viable for healthcare systems.
- Enhanced Supply Chain Reliability: The use of common chemical building blocks and standard reaction types ensures that raw materials are readily available from multiple suppliers. This reduces the risk of supply disruptions caused by shortages of specialized reagents. The scalability of the process means that production can be ramped up quickly to meet increasing demand as the drug candidate progresses through clinical trials. This flexibility is crucial for maintaining project timelines and ensuring that patient access to new treatments is not delayed by manufacturing bottlenecks. Reliable sourcing of these intermediates is key to a stable global supply chain.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing reaction conditions that can be safely translated from the laboratory to pilot and commercial scales. The reduction in hazardous waste generation aligns with modern green chemistry principles, facilitating easier regulatory approval and environmental compliance. Efficient solvent recovery systems can be integrated into the manufacturing process to further minimize environmental impact. This commitment to sustainability not only meets regulatory requirements but also enhances the corporate social responsibility profile of the manufacturing partner, appealing to ethically conscious stakeholders.
Frequently Asked Questions (FAQ)
The following questions address common inquiries regarding the technical and commercial aspects of these USP7 inhibitor intermediates. The answers are derived from the detailed technical disclosures within the patent documentation and reflect the current understanding of the chemistry involved. These insights are intended to assist decision-makers in evaluating the feasibility of incorporating these compounds into their development portfolios. Clear communication regarding technical capabilities and supply options is essential for fostering successful partnerships in the pharmaceutical industry.
Q: What is the primary therapeutic target of these intermediates?
A: These intermediates are designed to inhibit USP7 (Ubiquitin Specific Peptidase 7), a deubiquitinase involved in stabilizing oncoproteins like MDM2 and regulating p53 tumor suppressor pathways in various cancers.
Q: Are the synthesis routes scalable for commercial production?
A: Yes, the patented methods utilize standard organic transformations such as Suzuki coupling and nucleophilic substitution, which are well-established for scale-up from kilogram to multi-ton production levels.
Q: What purity levels can be expected from this synthesis?
A: The described purification methods, including flash column chromatography and recrystallization, are capable of achieving high-purity specifications suitable for preclinical and clinical grade material.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable USP7 Inhibitor Supplier
At NINGBO INNO PHARMCHEM, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and efficiency. Our team of expert chemists is well-versed in the complexities of heterocyclic synthesis and is equipped to handle the specific challenges associated with USP7 inhibitor intermediates. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch meets the highest standards of quality. Our commitment to technical excellence allows us to support your R&D efforts from the early discovery phase through to commercial manufacturing, providing a seamless transition as your program advances.
We invite you to contact our technical procurement team to discuss your specific requirements and explore how we can support your supply chain needs. Request a Customized Cost-Saving Analysis to understand how our optimized processes can benefit your project budget. We are ready to provide specific COA data and route feasibility assessments to help you make informed decisions. Partnering with us ensures access to high-purity USP7 inhibitors and a dedicated team focused on your success.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →