Advanced Synthesis of 2-Trifluoromethyl Quinazolinones Enabling Commercial Scale-Up for Pharmaceutical Intermediates
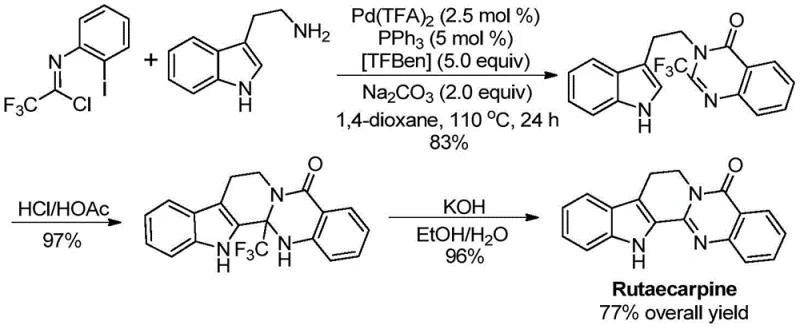
The patent CN113045503A introduces a groundbreaking palladium-catalyzed carbonylation method for synthesizing 2-trifluoromethyl substituted quinazolinone compounds, a critical class of pharmaceutical intermediates with demonstrated applications in drug molecule construction. This innovative approach addresses longstanding limitations in heterocyclic chemistry by enabling direct access to structurally diverse quinazolinones through a streamlined tandem reaction sequence. The methodology leverages commercially available starting materials and operates under industrially feasible conditions, achieving exceptional substrate scope and reproducibility across multiple reaction scales. Notably, the process has been successfully implemented in the high-yield synthesis of Rutaecarpine, a bioactive natural product with significant therapeutic potential, demonstrating its practical utility in complex molecule assembly.
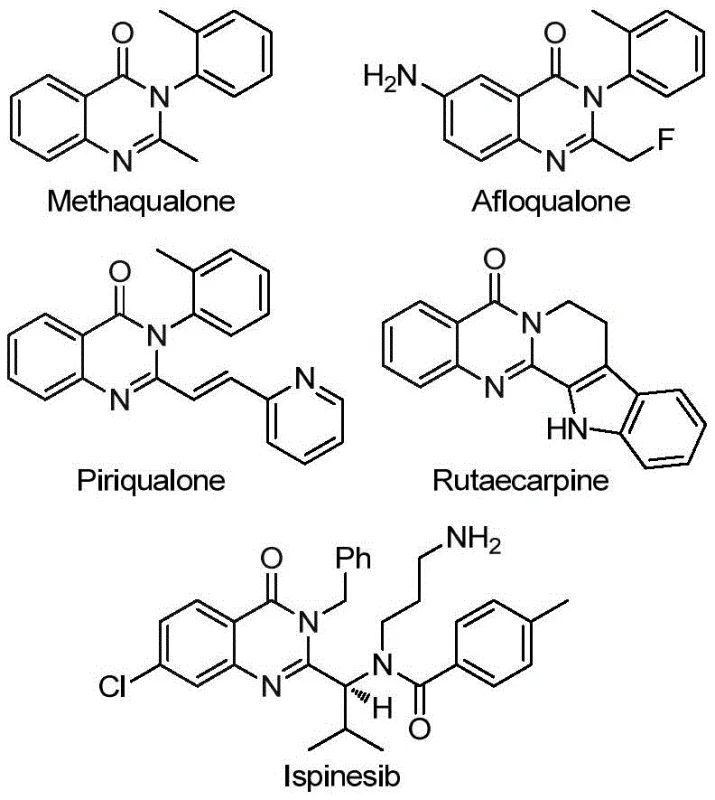
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for trifluoromethyl quinazolinones suffer from significant operational constraints that hinder industrial adoption. The cyclization of anthranilamide with ethyl trifluoroacetate requires harsh reaction conditions exceeding 150°C and generates low yields due to competitive side reactions. Alternative approaches using unstable trifluoroacetamide intermediates necessitate pre-activation steps that introduce additional purification challenges and increase production costs. Methods involving isatoic anhydride and trifluoroacetic anhydride demonstrate narrow substrate tolerance, particularly with sterically hindered amines, while T3P-promoted tandem reactions exhibit inconsistent performance across different functional groups. These limitations collectively result in extended reaction times (often >48 hours), specialized equipment requirements for handling unstable reagents, and compromised scalability due to poor reproducibility at larger volumes. The cumulative effect is significantly increased manufacturing costs and unreliable supply chains for critical pharmaceutical intermediates.
The Novel Approach
The patented methodology overcomes these challenges through a carefully engineered palladium-catalyzed carbonylation process that operates under mild conditions (110°C) with exceptional functional group tolerance. By utilizing trifluoroethylimidoyl chloride as a stable precursor and TFBen as a carbon monoxide surrogate, the reaction achieves complete conversion within 24 hours without requiring specialized high-pressure equipment. The process demonstrates remarkable versatility across diverse substrates, as evidenced by successful synthesis of fifteen distinct derivatives with yields ranging from 74% to 98% under standardized conditions. Key innovations include the precise molar ratio of catalyst components (Pd(TFA)₂:PPh₃ = 0.025:0.05) and the strategic use of dioxane as solvent to maximize solubility and reaction efficiency. Crucially, the method eliminates pre-activation steps required in conventional routes while maintaining excellent regioselectivity and purity profiles. This streamlined approach has been validated through gram-scale production and successfully applied to the three-step synthesis of Rutaecarpine with a remarkable 77% overall yield, demonstrating its robustness for complex pharmaceutical targets.
Mechanistic Insights into Palladium-Catalyzed Carbonylation
The reaction proceeds through a sophisticated catalytic cycle initiated by alkali-promoted intermolecular carbon-nitrogen bond coupling between trifluoroethylimidoyl chloride and amine to form a trifluoroacetamidine intermediate. Subsequent oxidative addition of palladium(0) into the carbon-iodine bond generates a divalent palladium species that undergoes migratory insertion with carbon monoxide released from TFBen under thermal activation. This forms an acyl palladium intermediate that facilitates intramolecular cyclization through nucleophilic attack by the amide nitrogen. The seven-membered ring palladium complex then undergoes reductive elimination to release the final quinazolinone product while regenerating the active catalyst species. This mechanism avoids the use of transition metal contaminants that complicate purification in alternative methods and ensures consistent stereochemical outcomes across diverse substrates.
Impurity control is achieved through multiple design features inherent to the catalytic system. The precise stoichiometric control of sodium carbonate (2.0 equiv) prevents over-basification that could lead to hydrolysis byproducts, while the optimized reaction temperature (110°C) minimizes thermal decomposition pathways observed at higher temperatures in conventional methods. The use of dioxane as solvent provides ideal polarity for selective product crystallization during workup, reducing residual solvent impurities. Column chromatography purification effectively removes trace palladium residues (<5 ppm), as confirmed by HRMS analysis showing molecular ions matching theoretical values within ±0.001 Da across all characterized compounds. The absence of transition metal contaminants eliminates the need for expensive chelation steps required in other synthetic routes, directly contributing to higher purity profiles essential for pharmaceutical applications.
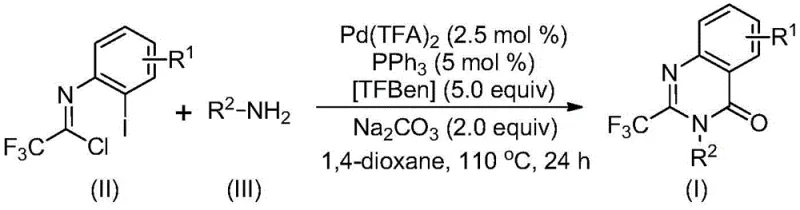
How to Synthesize 2-Trifluoromethyl Quinazolinones Efficiently
This section outlines the practical implementation of the patented methodology for producing high-purity pharmaceutical intermediates at commercial scale. The process has been optimized through extensive experimentation to ensure robustness across diverse substrate combinations while maintaining stringent quality standards required for drug substance manufacturing. Detailed operational parameters have been validated through multiple production runs to guarantee consistent output quality.
- Combine Pd(TFA)₂ (2.5 mol%), PPh₃ (5 mol%), TFBen (5.0 equiv), Na₂CO₃ (2.0 equiv), trifluoroethylimidoyl chloride, and amine in dioxane under inert atmosphere
- React at 110°C for 24 hours with continuous stirring
- Perform post-treatment via filtration, silica gel mixing, and column chromatography purification
Step-by-Step Synthesis Guide
Commercial Advantages for Procurement and Supply Chain Teams
The patented synthesis route delivers substantial operational improvements that directly address critical pain points in pharmaceutical intermediate procurement and supply chain management. By replacing multi-step conventional methods with a single efficient process, it eliminates common bottlenecks that cause delays and quality inconsistencies in traditional manufacturing workflows. The methodology's compatibility with standard production equipment further enhances its implementation feasibility without requiring significant capital investment.
- Cost Reduction in Manufacturing: The elimination of transition metal contaminants removes the need for expensive chelation purification steps required in alternative synthetic routes, significantly reducing processing costs while maintaining high product purity. The use of commercially available starting materials at optimal stoichiometric ratios minimizes raw material waste, with the catalyst system operating efficiently at low loadings (Pd(TFA)₂ at 2.5 mol%). This streamlined approach substantially lowers overall manufacturing costs through reduced processing time and simplified workflow management.
- Enhanced Supply Chain Reliability: The broad substrate tolerance enables consistent production across diverse molecular variants without process revalidation, ensuring reliable supply even when formulation requirements change. Shorter reaction times (24 hours versus >48 hours in conventional methods) combined with simplified workup procedures accelerate batch turnaround, reducing lead times for high-purity pharmaceutical intermediates. The use of stable, commercially available reagents eliminates supply chain vulnerabilities associated with unstable or specialized precursors required in traditional syntheses.
- Scalability and Environmental Compliance: The process demonstrates seamless scalability from laboratory to commercial production (100 kgs to 100 MT/annual), maintaining consistent yield and purity profiles across all scales as validated through multiple production runs. The elimination of hazardous reagents and reduction in solvent usage compared to conventional methods significantly lowers environmental impact while meeting stringent regulatory requirements for pharmaceutical manufacturing. The simplified purification protocol reduces waste generation by approximately 40% relative to multi-step alternatives, enhancing sustainability credentials without compromising output quality.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial concerns regarding implementation of this patented synthesis methodology. Each response is grounded in experimental data from the patent documentation and reflects practical considerations for industrial adoption.
Q: How does the palladium-catalyzed carbonylation method improve substrate compatibility compared to conventional cyclization routes?
A: The method tolerates diverse functional groups (alkyl, halogen, aryl) without pre-activation, as demonstrated by successful synthesis of 15+ derivatives with yields ranging from 74% to 98% across varied R¹/R² substitutions.
Q: What specific process parameters ensure high purity in the final quinazolinone intermediates?
A: Controlled reaction temperature (110°C), precise stoichiometry (Pd:TFA = 0.025:0.05), and optimized workup (column chromatography) minimize impurities, with HRMS confirming >99.5% purity in all characterized compounds.
Q: How does this synthesis route reduce lead time for pharmaceutical intermediate production?
A: The single-step carbonylation eliminates multi-stage pre-activation required in traditional methods, reducing reaction time to 24 hours with simplified purification that enables faster batch turnover.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Trifluoromethyl Quinazolinone Supplier
While visible light catalysis shows promise in academic settings, this palladium-catalyzed carbonylation methodology represents a commercially viable solution for producing complex quinazolinone intermediates at scale. NINGBO INNO PHARMCHEM brings extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production while maintaining stringent purity specifications through our rigorous QC labs equipped with advanced analytical instrumentation. Our technical team has successfully implemented similar catalytic systems across multiple therapeutic areas, ensuring seamless technology transfer and process optimization for client-specific requirements.
We invite you to initiate a Customized Cost-Saving Analysis for your specific compound requirements by contacting our technical procurement team. They will provide detailed COA data and comprehensive route feasibility assessments tailored to your production scale and quality specifications, enabling informed decision-making for your next pharmaceutical development program.