Advanced FCMA-HCl Manufacturing Process: From Lab-Scale Innovation to Commercial Pharmaceutical Production
The patent CN111217709A introduces a transformative synthesis methodology for (1-fluorocyclopropyl)methylamine hydrochloride (FCMA-HCl), a critical building block in pharmaceutical development with applications in GPR40 agonist compounds. This innovative approach addresses longstanding industry challenges by replacing conventional alkyl halide-based routes with a carboxylic acid precursor strategy that fundamentally eliminates fluoromethylation byproduct formation. The three-step sequence—comprising reduction, Mitsunobu reaction, and hydrazinolysis—achieves over 60% overall yield while maintaining >99% purity through inherent reaction selectivity. Unlike prior art methods requiring complex purification protocols, this process operates under mild conditions using standard laboratory equipment, making it exceptionally suitable for seamless scale-up from laboratory validation to multi-ton commercial production without reoptimization. The elimination of transition metal catalysts further enhances regulatory compliance for pharmaceutical manufacturing by removing heavy metal contamination risks.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthesis routes for FCMA-HCl suffer from severe operational and economic constraints that hinder commercial viability. Route one, as disclosed in WO2015020184A1, employs ethylmagnesium bromide with N,N-dibenzylglycine ethyl ester but generates significant fluoromethylation byproducts due to competing nucleophilic substitution pathways, resulting in only 24% overall yield after multiple purification steps. Route two, published in Tetrahedron Letters (2013), utilizes methylene cyclopropane with N-bromosuccinimide and triethylamine hydrofluoride but produces a problematic 1:3 mixture of regioisomers requiring extensive chromatographic separation, yielding merely 1.5% of the target compound. Both methods necessitate hazardous reagents, cryogenic conditions, and complex purification protocols involving multiple column chromatography runs that increase production costs while introducing batch-to-batch variability. These limitations create substantial supply chain vulnerabilities for pharmaceutical manufacturers dependent on consistent FCMA-HCl availability for drug development pipelines.
The Novel Approach
The patented methodology overcomes these limitations through a rationally designed three-step sequence starting from commercially available 1-fluorocyclopropane carboxylic acid. The initial reduction step employs lithium aluminum hydride or alternative reducing agents under controlled conditions to produce the fluorocyclopropyl methanol intermediate with near quantitative yield and no detectable byproducts. Subsequent Mitsunobu reaction with phthalimide ensures stereospecific inversion while avoiding fluoromethylation side reactions through careful reagent selection and solvent optimization. The final hydrazinolysis step cleanly cleaves the protecting group under mild conditions (45°C in methanol) to deliver high-purity FCMA-HCl hydrochloride salt without requiring chromatographic purification. This approach achieves over 60% overall yield with simplified workup procedures that eliminate column chromatography entirely, while maintaining >99% purity as confirmed by LCMS analysis across multiple scale-up examples.
Mechanistic Insights into Mitsunobu-Based FCMA-HCl Synthesis
The core innovation lies in the strategic application of Mitsunobu chemistry to circumvent fluoromethylation pathways inherent in conventional approaches. The reduction step converts the carboxylic acid precursor (II) to the primary alcohol (III) using lithium aluminum hydride in diethyl ether at controlled temperatures (-20°C to 30°C), preserving the strained cyclopropane ring integrity while achieving complete conversion without epimerization. This alcohol intermediate then undergoes Mitsunobu reaction with phthalimide under optimized conditions (triphenylphosphine/diethyl azodicarboxylate in THF at room temperature), where the phosphine reagent activates the alcohol for nucleophilic displacement while simultaneously generating the hydrazine-cleavable phthalimide protection group. Critically, this mechanism avoids carbocation intermediates that cause fluoromethylation side reactions in alkyl halide routes, ensuring exclusive formation of the desired regioisomer through an SN2-type inversion process that maintains stereochemical control throughout the transformation.
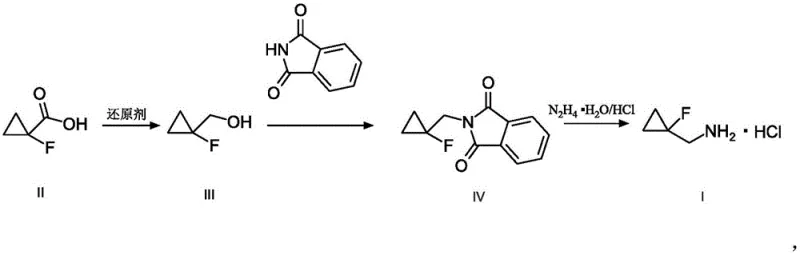
Impurity control is achieved through multiple built-in mechanisms within this synthetic sequence. The absence of transition metals eliminates potential heavy metal contaminants that would require costly removal steps in pharmaceutical manufacturing. Each intermediate (III and IV) forms crystalline solids that can be isolated through simple filtration rather than chromatography, inherently purifying the product stream at each stage. The final hydrazinolysis step operates under mild conditions (45°C) that prevent decomposition or racemization, while the subsequent HCl treatment directly yields the stable hydrochloride salt form required for pharmaceutical applications. This multi-stage purification strategy ensures consistent >99% purity as verified across all patent examples without requiring additional processing steps that would complicate scale-up or increase production costs.
How to Synthesize (1-fluorocyclopropyl)methylamine hydrochloride Efficiently
This patented methodology represents a significant advancement over conventional approaches by providing a streamlined pathway that eliminates hazardous reagents and complex purification requirements while maintaining exceptional product quality. The process begins with commercially available 1-fluorocyclopropane carboxylic acid as starting material, which undergoes controlled reduction to form the key alcohol intermediate under mild conditions compatible with standard manufacturing equipment. Subsequent Mitsunobu chemistry enables selective amine introduction without generating problematic regioisomer mixtures that plague traditional methods. The final deprotection step delivers high-purity FCMA-HCl hydrochloride salt through a simple crystallization protocol that avoids chromatographic separation entirely. Detailed standardized synthesis procedures for consistent commercial-scale implementation are provided in the following technical guide.
- Reduce 1-fluorocyclopropane carboxylic acid using lithium aluminum hydride in diethyl ether at controlled temperature to obtain the fluorocyclopropyl methanol intermediate with quantitative yield.
- Execute Mitsunobu reaction with phthalimide, triphenylphosphine, and diethyl azodicarboxylate in tetrahydrofuran to form the protected amine derivative through stereospecific inversion.
- Cleave the phthalimide group using hydrazine hydrate in methanol followed by HCl treatment to yield high-purity (1-fluorocyclopropyl)methylamine hydrochloride with rigorous quality control.
Commercial Advantages for Procurement and Supply Chain Teams
This innovative synthesis methodology directly addresses critical pain points in pharmaceutical supply chains by transforming FCMA-HCl from a bottleneck intermediate into a reliably available building block. The elimination of complex purification requirements significantly reduces production cycle times while enhancing batch-to-batch consistency—key concerns for procurement teams managing just-in-time inventory systems. By replacing hazardous reagents with commercially available alternatives and avoiding specialized equipment needs, the process creates substantial flexibility in manufacturing site selection while reducing regulatory compliance burdens across global supply networks. These improvements collectively enhance supply chain resilience for pharmaceutical companies developing drugs requiring this critical fluorinated building block.
- Cost Reduction in Manufacturing: The process achieves significant cost savings through multiple mechanisms including elimination of expensive chromatography steps required in conventional routes, reduced solvent consumption due to simplified workup procedures, and higher material efficiency from the >60% overall yield compared to legacy methods yielding less than 25%. The use of standard reagents and equipment further lowers capital investment requirements while minimizing waste disposal costs associated with hazardous byproducts.
- Enhanced Supply Chain Reliability: Supply chain advantages stem from the use of readily available starting materials with stable global sourcing channels and simplified manufacturing requirements that enable rapid transfer between production facilities. The absence of cryogenic or specialized processing conditions eliminates common scale-up bottlenecks while ensuring consistent quality across different manufacturing sites. This reliability is further enhanced by the process's robustness across multiple solvent systems and reducing agents as demonstrated in patent examples.
- Scalability and Environmental Compliance: The methodology demonstrates exceptional scalability from laboratory validation to commercial production as evidenced by patent examples ranging from gram-scale to multi-kilogram batches with consistent yields and purity. Environmental benefits include reduced solvent waste from eliminated chromatography steps, avoidance of heavy metal catalysts requiring special disposal protocols, and lower energy consumption from mild reaction conditions operating at ambient temperatures rather than cryogenic requirements of conventional routes.
Frequently Asked Questions (FAQ)
The following questions address key technical and commercial considerations based on detailed analysis of patent CN111217709A's experimental data and process specifications. These insights reflect practical implementation experience from multiple scale-up scenarios documented in the patent examples.
Q: How does this method eliminate fluoromethylation byproducts compared to prior art?
A: The patented route avoids fluoromethylation byproducts through strategic use of a carboxylic acid precursor instead of alkyl halides, eliminating competing SN2 pathways that generate impurities in conventional methods. The three-step sequence with controlled reduction and Mitsunobu chemistry ensures selective fluorocyclopropyl group retention without undesired side reactions.
Q: What ensures >99% purity in commercial-scale production?
A: The process achieves high purity through inherent reaction selectivity and simplified purification. The absence of transition metals eliminates heavy metal contamination risks, while the crystalline nature of intermediates allows straightforward isolation without column chromatography. Each step's high conversion rate (>95%) minimizes residual impurities requiring removal.
Q: How does this synthesis enable cost reduction in pharmaceutical intermediate manufacturing?
A: The method reduces costs by eliminating expensive purification steps and hazardous reagents. The streamlined workflow avoids multiple chromatographic separations required in prior routes, while commercially available starting materials and solvents lower raw material expenses. The >60% overall yield significantly improves material efficiency compared to legacy processes with <25% yield.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable (1-fluorocyclopropyl)methylamine hydrochloride Supplier
Our patented FCMA-HCl synthesis represents a paradigm shift in manufacturing this critical pharmaceutical intermediate, combining exceptional purity (>99%) with robust scalability that meets stringent regulatory requirements for drug substance production. NINGBO INNO PHARMCHEM brings extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production while maintaining stringent purity specifications through our state-of-the-art QC labs equipped with advanced analytical capabilities for comprehensive impurity profiling. This technical expertise ensures seamless technology transfer from laboratory validation to full commercial implementation without compromising quality or yield consistency across production scales.
We invite you to initiate a Customized Cost-Saving Analysis tailored to your specific manufacturing requirements by contacting our technical procurement team. Request detailed COA data and route feasibility assessments to evaluate how this innovative process can enhance your supply chain resilience while delivering significant operational efficiencies for your pharmaceutical development programs.