Advanced Manufacturing of (1-Fluorocyclopropyl)methylamine Hydrochloride for Pharmaceutical Intermediates
Advanced Manufacturing of (1-Fluorocyclopropyl)methylamine Hydrochloride for Pharmaceutical Intermediates
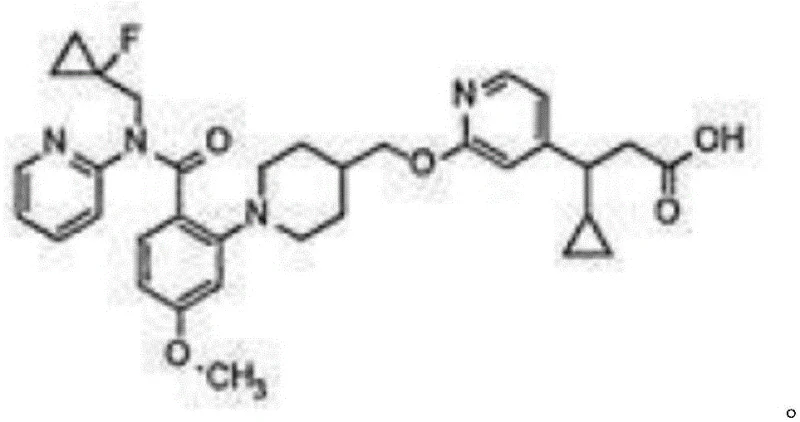
The pharmaceutical industry continuously demands high-purity building blocks for the synthesis of complex therapeutic agents, particularly those targeting metabolic disorders. Patent CN111217709A introduces a groundbreaking preparation method for (1-fluorocyclopropyl)methylamine hydrochloride, a critical intermediate utilized in the development of GPR40 agonists. This technical disclosure outlines a robust three-step synthetic pathway starting from 1-fluorocyclopropanecarboxylic acid, offering a substantial improvement over legacy manufacturing techniques. By leveraging a sequence of reduction, ammoniation via Mitsunobu reaction, and deprotection, the process achieves a total reaction yield exceeding 60 percent. This represents a paradigm shift for procurement teams seeking reliable pharmaceutical intermediate suppliers who can guarantee both cost efficiency and structural integrity in their supply chains.
For R&D directors evaluating process feasibility, the significance of this patent lies in its ability to bypass the notorious impurity profiles associated with fluorinated cyclopropyl amines. The introduction of fluorine atoms into lead compounds is a common strategy to enhance lipophilicity and metabolic stability, yet the synthesis of such motifs often suffers from low atom economy. The method described in CN111217709A addresses these challenges directly by utilizing mild reaction conditions and readily available reagents. As a result, the final product demonstrates exceptional purity levels, often reaching 99 percent as confirmed by LCMS analysis, thereby reducing the burden on downstream purification processes and ensuring consistent quality for clinical trial materials.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this innovation, the synthesis of (1-fluorocyclopropyl)methylamine hydrochloride was plagued by significant technical hurdles that hindered commercial viability. International patent WO2015020184a1 disclosed a route initiating from ethylmagnesium bromide and N,N-dibenzylglycine ethyl ester, which unfortunately resulted in a meager total yield of approximately 24 percent. Furthermore, academic literature such as Tetrahedron Letters (2013) described an alternative pathway using methylenecyclopropane and N-bromosuccinimide, which performed even worse with a yield of merely 1.5 percent. These conventional methods inevitably produce significant quantities of fluoromethylation by-products, creating a complex mixture that requires multiple column chromatography purifications to isolate the target amine.
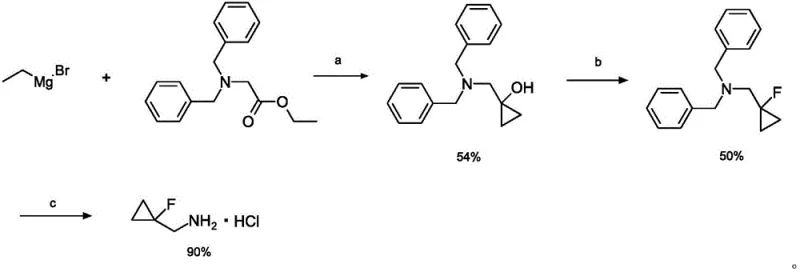
The presence of these difficult-to-remove impurities not only drives up manufacturing costs but also poses risks to the safety profile of the final API. The reliance on harsh fluorinating agents and the generation of isomeric by-products, such as the 1:3 ratio of regioisomers observed in older routes, complicates the regulatory filing process. For supply chain heads, the low throughput and extensive purification requirements of these legacy methods translate into long lead times and unpredictable availability. Consequently, there has been an urgent need within the fine chemical sector for a streamlined approach that eliminates these bottlenecks while maintaining high stereochemical and chemical purity standards.
The Novel Approach
The novel approach detailed in CN111217709A offers a decisive solution by restructuring the synthetic logic to avoid fluoromethylation by-products entirely. Instead of attempting to introduce fluorine late in the sequence or via unstable intermediates, this method starts with 1-fluorocyclopropanecarboxylic acid, a stable and commercially accessible feedstock. The core transformation involves converting the carboxylic acid into the corresponding primary alcohol, followed by a nitrogen installation step using phthalimide, and finally releasing the free amine. This strategic redesign ensures that the fluorine atom remains intact on the cyclopropyl ring throughout the synthesis, preventing the scrambling and side reactions that characterize earlier attempts.
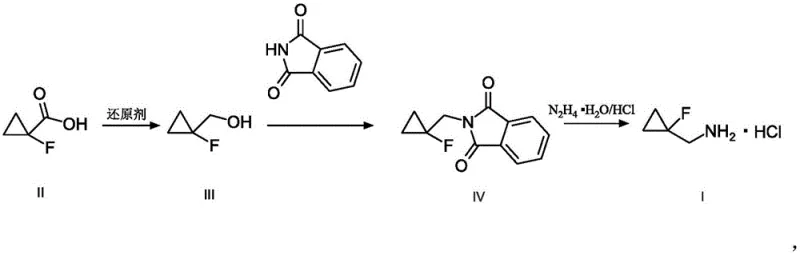
By implementing this route, manufacturers can achieve a reaction yield of more than 60 percent across three steps, a dramatic improvement over the single-digit or low-double-digit yields of the past. The operational simplicity is another key advantage; the process avoids the need for complex cryogenic conditions or exotic catalysts, relying instead on standard unit operations like filtration and extraction. This makes the technology highly attractive for cost reduction in pharmaceutical intermediate manufacturing, as it allows for scaling from kilogram to multi-ton quantities without the exponential cost increases typically associated with chromatographic purification. The result is a high-purity product that meets stringent specifications with minimal environmental waste.
Mechanistic Insights into the Three-Step Synthetic Sequence
The success of this preparation method relies on the precise optimization of three distinct chemical transformations, each selected for its robustness and scalability. The first step involves the reduction of 1-fluorocyclopropanecarboxylic acid to (1-fluorocyclopropyl)methanol. The patent explores various reducing agents, including lithium aluminum hydride, sodium borohydride combined with boron trifluoride etherate, and borane complexes. Mechanistically, the hydride source attacks the carbonyl carbon of the carboxylic acid, eventually leading to the primary alcohol. The choice of solvent, such as diethyl ether or tetrahydrofuran, is critical to manage the exothermicity of the reaction and ensure complete conversion without opening the strained cyclopropane ring.
Following the reduction, the second step employs a classic Mitsunobu reaction to install the nitrogen functionality. In this transformation, the newly formed alcohol reacts with phthalimide in the presence of triphenylphosphine and an azo compound, such as diethyl azodicarboxylate (DEAD) or diisopropyl azodicarboxylate (DIAD). This step proceeds via the formation of an alkoxyphosphonium ion, which is then displaced by the phthalimide anion. This mechanism is particularly advantageous because it proceeds under mild conditions and avoids the use of harsh alkylating agents that could compromise the fluorinated cyclopropyl motif. The resulting N-substituted phthalimide intermediate is a stable solid that can be easily purified by crystallization or simple washing, effectively locking in the purity before the final step.
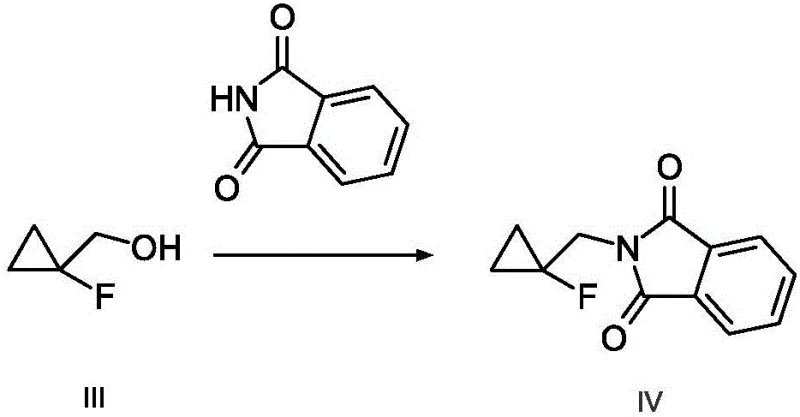
The final stage of the synthesis is the deprotection of the phthalimide group to release the free amine, which is subsequently converted to its hydrochloride salt. This is achieved through hydrazinolysis, where hydrazine hydrate reacts with the imide to form phthalhydrazide and the free amine. The reaction is typically conducted in methanol or ethanol at moderate temperatures ranging from 30 to 60 degrees Celsius. The addition of hydrogen chloride in the final workup ensures the precipitation of the amine as a stable hydrochloride salt. This sequence effectively controls the impurity profile by ensuring that any unreacted intermediates or side products are removed during the aqueous workups, resulting in a final product with purity levels suitable for direct use in API synthesis without further chromatography.
How to Synthesize (1-Fluorocyclopropyl)methylamine Hydrochloride Efficiently
Implementing this synthesis route requires careful attention to stoichiometry and temperature control to maximize the impressive yields reported in the patent data. The process begins with the activation of the reducing agent, followed by the controlled addition of the fluoro-acid substrate to prevent thermal runaway. Subsequent steps involve the precise mixing of phosphine and azo reagents to drive the Mitsunobu coupling to completion. While the general chemistry is straightforward, the specific molar ratios and solvent choices outlined in the examples are crucial for reproducing the >60 percent overall yield. For detailed operational parameters, please refer to the standardized guide below.
- Reduce 1-fluorocyclopropanecarboxylic acid to (1-fluorocyclopropyl)methanol using lithium aluminum hydride or borane complexes in ether solvents.
- Perform a Mitsunobu reaction between the resulting alcohol and phthalimide using triphenylphosphine and an azo compound like diethyl azodicarboxylate.
- Cleave the phthalimide protecting group using hydrazine hydrate followed by acidification with hydrogen chloride to obtain the final amine salt.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this novel synthesis route offers transformative benefits for procurement managers and supply chain strategists looking to optimize their sourcing of fluorinated intermediates. The primary driver of value is the drastic simplification of the purification workflow. By eliminating the need for multiple column chromatography separations, which are notoriously expensive and difficult to scale, the process significantly reduces the consumption of silica gel and organic solvents. This reduction in material usage directly translates to lower variable costs per kilogram of product, allowing for more competitive pricing structures in long-term supply agreements without sacrificing margin.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the avoidance of complex purification steps create a leaner manufacturing cost structure. Traditional routes often require specialized equipment for handling hazardous fluorinating agents and extensive downtime for column packing and running. In contrast, this new method relies on commodity chemicals like phthalimide and hydrazine, which are available in bulk at stable prices. Furthermore, the high yield means that less raw material is wasted, improving the overall atom economy and reducing the cost of goods sold substantially for large volume contracts.
- Enhanced Supply Chain Reliability: Supply continuity is often threatened by the reliance on custom-synthesized starting materials or reagents with long lead times. This process utilizes 1-fluorocyclopropanecarboxylic acid, which is becoming increasingly available from global chemical suppliers, thereby diversifying the supply base. The robustness of the reaction conditions also means that production is less susceptible to batch failures caused by minor fluctuations in temperature or moisture. For supply chain heads, this reliability ensures that inventory levels can be maintained more predictably, reducing the risk of stockouts that could delay critical drug development timelines.
- Scalability and Environmental Compliance: As the pharmaceutical industry moves towards greener manufacturing practices, the environmental footprint of intermediate synthesis is under increasing scrutiny. This route generates fewer hazardous by-products compared to the bromination-fluorination sequences of the past. The waste streams are primarily composed of triphenylphosphine oxide and phthalhydrazide, which are easier to manage and dispose of than heavy metal residues or fluorinated organic waste. This facilitates easier regulatory compliance and permits faster scale-up from pilot plant to commercial production scales, ensuring that the supply chain can respond agilely to market demand surges.
Frequently Asked Questions (FAQ)
Understanding the technical nuances of this synthesis is vital for stakeholders evaluating its integration into their supply networks. The following questions address common concerns regarding yield consistency, impurity profiles, and scalability. These answers are derived directly from the experimental data provided in the patent documentation, ensuring that the information is grounded in verified laboratory results rather than theoretical projections. This transparency helps build trust between suppliers and pharmaceutical partners.
Q: How does the new synthesis route compare to prior art in terms of yield?
A: The novel three-step route achieves a total yield of over 60%, significantly outperforming previous methods which reported yields of only 24% and 1.5% respectively due to byproduct formation.
Q: What are the key impurities avoided in this manufacturing process?
A: This method specifically avoids the generation of fluoromethylation by-products that are inevitable in conventional Grignard or halogenation-fluorination routes, simplifying purification.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the process utilizes commercially available starting materials and standard reagents like LiAlH4 and phthalimide, avoiding complex column chromatography, making it highly scalable.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable (1-Fluorocyclopropyl)methylamine Hydrochloride Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of novel therapeutics depends on the availability of high-quality intermediates produced via robust pathways. Our team of process chemists has extensively analyzed the technology disclosed in CN111217709A and is fully prepared to implement this superior three-step synthesis at scale. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and speed. Our facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of (1-fluorocyclopropyl)methylamine hydrochloride meets the exacting standards required for GPR40 agonist development.
We invite you to collaborate with us to leverage this advanced manufacturing capability for your next project. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis that quantifies the economic benefits of switching to this high-yield route. We encourage you to contact us today to request specific COA data and route feasibility assessments tailored to your unique volume requirements. Let us help you secure a stable, cost-effective supply of this critical fluorinated building block, enabling you to focus on what matters most: delivering life-saving medicines to patients worldwide.